| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
EGFR (IC50 = 3 nM); HCV; EMCV
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
在化学指定的 DMEM/F12 培养基中,培养人肺癌 (A549) 和前列腺 (DU145) 癌细胞系。 AG-1478 (AG1478) 对这些细胞系的生长调节是不可逆的。尽管 AG-1478 不能完全阻止 A549 细胞的增殖,但它在较低浓度下似乎更有效[1]。当 EGFR 被特异性酪氨酸激酶抑制剂 AG-1478 (AG1478) 抑制时,血管紧张素 II 介导的心脏成纤维细胞产生的 TGF-β 和纤连蛋白会显着减少。 AG-1478 是一种 IC50 为 4 nM 的小分子抑制剂,在药理学上抑制 EGFR[2]。使用流式细胞术确定 Polyfect (PF) 和 Superfect (SF) 处理对 HEK 293 细胞凋亡的促进程度。 Tempol 是一种抗氧化剂,可显着降低 PF 和 SF 树枝状聚合物介导的细胞凋亡。作为阳性对照,AG-1478(AG1478)的使用剂量比信号研究的剂量高10倍(100μM),它有效地引起HEK 293细胞凋亡[3]。
体外活性:AG-1478(也称为 Tyrphostin AG-1478)是一种有效的选择性 EGFR(表皮生长因子受体)抑制剂,在无细胞测定中 IC50 为 3 nM。它可逆地抑制大鼠脑 Kv1.5 钾通道,IC50 为 9.8 µM,与蛋白酪氨酸激酶 (PTK) 活性无关。 AG-1478 还抑制平滑肌瘤和子宫肌层细胞培养物的生长,IC50 值分别为 5.6 和 5.7 µM。此前的研究表明,EGFR拮抗作用可能有效治疗多种疾病,如癌症、血管紧张素II诱导的心脏肥大和糖尿病心肌病。因此,AG-1478 有潜力用作这些疾病的治疗药物。激酶测定:AG-1478 对 ErbB2 和 PDGFR 具有高选择性,IC50 > 100 μM。与表达内源wt EGFR或过表达外源wt EGFR的细胞相比,AG-1478优先抑制表达截短EGFR的U87MG细胞,IC50为8.7μM,IC50分别为34.6μM和48.4μM,并抑制DNA合成,IC50为4.6μM,分别为 19.67 μM 和 35.2 μM。与内源性或过表达的外源性 wt EGFR 相比,AG-1478 还优先抑制 ΔEGFR 的酪氨酸激酶活性和自身磷酸化。 AG-1478 (0.25 μM) 消除由 Ang II(一种 Ca2+ 离子载体)以及 EGF 诱导的 MAPK 激活,但不能消除 VSMC 中佛波醇酯或血小板衍生生长因子-BB 诱导的 MAPK 激活。 AG-1478 抑制 EGF 诱导的 BaF/ERX 和 LIM1215 细胞有丝分裂,IC50 分别为 0.07 μM 和 0.2 μM。 AG1478 能够抑制 ATP 结合盒 (ABC) 转运蛋白(例如 ABCB1 和 ABCG2)的功能,其中对 ABCG2 的影响更为明显。细胞测定:将细胞在 96 孔板中暴露于不同浓度的 AG-1478 中 72 小时。使用 Alamar Blue 测定法检查 AG-1478 对细胞生长的影响。将 20 μL 等份的 Alamar Blue 添加到每个孔中,并使用 Spectromax 扫描微孔板读数仪测定其吸光度。 AG-1478 的效果以生长抑制百分比表示,使用未处理的细胞作为对照(0% 抑制)。使用 [3H] 胸苷掺入测定法测定细胞 DNA 合成。 我们使用了两种选择性EGFR酪氨酸激酶抑制剂:AG494(可逆)和AG1478(不可逆),用于在化学定义的DMEM/F12培养基中培养的人肺(A549)和前列腺(DU145)癌症细胞系的生长调节。两种测试的酪氨酸磷酸酶都显著抑制了所研究细胞系的自分泌生长。AG494的作用呈剂量依赖性,在最高浓度下可完全抑制生长。AG1478在较低浓度下似乎更有效,但无法完全抑制A549细胞的生长。与AG1478相比,AG494对EGFR激酶活性的抑制对两种细胞系中ERK的活性没有影响。两种EGFR抑制剂都诱导了所研究的肺癌和前列腺癌癌症细胞系的凋亡,但所研究的tyrphostins在A549中的促凋亡作用大于在DU145细胞中的促细胞凋亡作用。与其他已知的EGFR抑制剂类似,酪氨酸磷酸酶在G1期抑制了DU145和A549细胞的生长。AG494和AG1478对两种信号蛋白(AKT和ERK)活性的影响取决于所研究的细胞类型。在DU145细胞的情况下,两种激酶的酶活性都明显下降(AG1478更强),而在A549中,只有AG1478有效地抑制了Akt的磷酸化。Tyrphostins AG494和AG1478是ATP竞争对手,应该具有类似的作用机制,但我们的研究结果表明这并不完全正确。[1] EGFR抑制剂预防H9C2细胞PA诱导的损伤[2] 为了评估EGFR抑制剂对体外心肌细胞肥大的影响,H9C2细胞用AG1478或542预处理2小时,然后暴露于PA(100μM)6小时。使用罗丹明标记的Phalloidin评估细胞形态和细胞体积。PA的孵育显著增加了H9C2细胞的体积,而AG或542可以逆转细胞形态的变化(图6A,B)。同时,AG或542预处理显著降低了H9C2细胞中肥大标志物基因心钠素(ANP)和促纤维化基因TGF-β的mRNA水平(图6C,D),表明这些小分子抑制剂可以预防PA诱导的心肌细胞肥大和纤维化。 基因敲除EGFR抑制的PA诱导的H9C2细胞炎症损伤[2] 为了避免小分子抑制剂的非特异性并验证EGFR的作用,我们用特异性小干扰RNA转染H9C2细胞以下调EGFR表达(si-EGFR)。如图7A所示,在100μM PA处理下转染si-EGFR显著降低了H9C2细胞中EGFR蛋白的表达,进一步导致TNF-α和ANP基因表达水平降低,PA刺激的H9C2细胞中caspase-3活性降低(图7B-D)。这些结果,再加上我们对542/AG1478细胞内效应的观察,证实了EGFR在介导高脂血症诱导的心脏损伤中起着重要作用。为了模拟临床环境,我们进一步评估了EGFR抑制对已经暴露于PA(作为治疗)的H9c2细胞中PA有害作用的保护作用。补充图S3中的结果显示,用EGFR抑制剂AG或542治疗后,PA也降低,TNF-α和ANP水平升高。 脑心肌炎病毒(EMCV)与丙型肝炎病毒(HCV)一样,需要磷脂酰肌醇4-激酶IIIα(PI4KA)进行基因组复制。在这里,我们证明了已知的表皮生长因子受体(EGFR)抑制剂tyrphostinAG1478也在体外和细胞内抑制PI4KA活性。AG1478会损害EMCV和HCV的复制,但不会损害先前证明可以逃避PI4KA抑制的EMCV突变体的复制。这项工作揭示了AG1478的新细胞和抗病毒特性,AG1478是一种以前仅被视为癌症化疗剂的化合物[4]。 |
||
| 体内研究 (In Vivo) |
在这两种肥胖小鼠模型中,给予 AG-1478 (AG1478) 可显着降低心肌炎症、纤维化、细胞凋亡和功能障碍。 ApoE-/- 小鼠在喂食 HFD 8 周(ApoE-HFD)后,口服 AG -1478(10 mg/kg/天)或 542(10 mg/kg/天)灌胃 8 周。在体内,AG-1478 或 542 疗法可阻断 HFD 诱导的心脏 EGFR 磷酸化,但对血浆总甘油三酯 (TG) 或低密度脂蛋白 (LDL) 水平没有影响[2]。施用 EGF (10 nM) 后,EGFR 磷酸化持续强烈升高,并且这种升高可以被已知的 EGFR 磷酸化抑制剂 AG-1478 (AG478) 抑制。尽管不如 AG1478 那么多,但增加 Polyfect (PF) 剂量可显着降低 EGF 诱导的 EGFR 磷酸化 (p<0.05)[3]。
AG-1478的施用可阻断肿瘤部位EGFR的磷酸化,并抑制过表达WT EGFR的A431异种移植物和表达de2-7 EGFR的神经胶质瘤异种移植物的生长。即使是亚治疗剂量的 AG-1478 也能显着增强细胞毒性药物的功效,AG-1478 和替莫唑胺的组合对人神经胶质瘤异种移植物表现出协同抗肿瘤活性。 AG-1478 和抗 EGFR 抗体 (mAb 806) 的组合对过度表达 EGFR 的肿瘤异种移植物表现出相加的、在某些情况下协同的抗肿瘤活性。 AG-1478(0.4 mg)与单剂量 25 μCi 90Y-CHX-A-DTPA-hu3S193 的组合与单独使用任一药物相比,可显着增强疗效。 使用喂食高脂肪饮食(HFD)的野生型(WT)和载脂蛋白E(ApoE)敲除小鼠的体内研究表明,小分子EGFR抑制剂AG1478和542对肥胖诱导的心肌损伤具有有益作用。AG1478和542的给药显著降低了两种肥胖小鼠模型的心肌炎症、纤维化、细胞凋亡和功能障碍。在体外,棕榈酸(PA)刺激的心肌细胞中,EGFR信号传导被siRNA沉默或小分子EGFR抑制剂阻断。EGFR抑制减弱了PA诱导的H9C2细胞炎症反应和凋亡。此外,我们发现PA诱导的EGFR激活是由上游TLR4和c-Src介导的。本研究证实了EGFR活化在实验小鼠肥胖诱导的心脏炎症损伤发病机制中的有害作用,并证明了TLR4/c-Src介导的PA诱导EGFR活化机制。我们的数据表明,EGFR可能是肥胖相关心血管疾病的治疗靶点[2]。 |
||
| 酶活实验 |
AG-1478 的 IC50 > 100 μM,对 ErbB2 和 PDGFR 具有高度选择性。在 U87MG 细胞中,AG-1478 优先抑制截短 EGFR 表达 (IC50 = 8.7 μM),而不是内源 wt EGFR 表达(IC50 = 34.6 μM 和 48.4 μM,分别),并抑制 DNA 合成(IC50 = 4.6 μM、19.67 μM 和 35.2)分别为μM)。此外,与内源性或过表达的外源性 wt EGFR 相比,AG-1478 优先抑制 ΔEGFR 的酪氨酸激酶活性和自身磷酸化。在 VSMC 中,AG-1478 (0.25 μM) 可消除由 Ang II(一种 Ca2+ 离子载体)和 EGF 引起的 MAPK 激活,但不能消除由佛波醇酯或血小板衍生生长因子-BB 引起的 MAPK 激活。 AG-1478 的 IC50 值分别为 0.07 μM 和 0.2 μM,抑制 EGF 诱导的 BaF/ERX 和 LIM1215 细胞有丝分裂。 AG1478 可抑制 ATP 结合盒 (ABC) 转运蛋白 ABCB1 和 ABCG2,其中对 ABCG2 的影响更大。
|
||
| 细胞实验 |
A549 (CCL-185) 和 DU145 (HTB-81) 细胞以 4×103 细胞/孔的密度接种在 DMEM(A549 细胞)或 MEM(DU145 细胞)中的 96-孔板。 24 小时孵育期后,用白蛋白 (0.5 mg/mL)、亚硒酸钠 (2 ng/mL) 和转铁蛋白 (5 mg/mL) 增强的无血清 DMEM/F12 (1:1) 替代培养基(DMEM/F12+)。孵育期第 0 天后,用含有浓度分别为 1-20 μM 和 0.1-8 μM 的酪氨酸激酶抑制剂(AG494、AG-1478)的无血清 DMEM/F12+ 更换培养基。在接下来的 24 小时内,孵化保持在 37°C 的潮湿环境中。使用 MTT 测定和改良的结晶紫染色法 (CV) 评估酪氨酸磷酸酶对靶细胞增殖的影响。 Tecan 多重扫描平板记录仪用于测量吸光度[1]。
|
||
| 动物实验 |
|
||
| 参考文献 |
|
||
| 其他信息 |
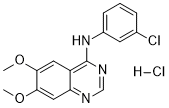
Tyrphostin AG1478 属于喹唑啉类化合物,其喹唑啉环在 6 位和 7 位被甲氧基取代,4 位被 (3-氯苯基)亚硝基取代。它是一种表皮生长因子受体拮抗剂。Tyrphostin AG1478 具有多种药理活性,包括表皮生长因子受体拮抗剂、抗肿瘤药、抗衰老药和抗病毒药。它属于喹唑啉类、芳香醚类和一氯苯类化合物。Tyrphostin AG1478 是酪氨酸激酶抑制剂 Tyrphostin 家族的成员,可选择性抑制表皮生长因子。阳离子聚酰胺胺 (PAMAM) 树状聚合物是一类分支状球形聚合物,目前正在研究其在纳米医学领域的多种应用,包括核酸药物递送。新出现的证据表明,它们具有内在的生物学和毒理学效应,但人们对它们与信号转导通路的相互作用知之甚少。我们之前已证明,活化的(片段化的)第6代(G)PAMAM树状聚合物Superfect(SF)能够刺激培养的人胚肾(HEK 293)细胞中的表皮生长因子受体(EGFR)酪氨酸激酶信号传导——这是一个重要的信号级联,可调节细胞生长、存活和凋亡。本研究首先探讨了非活化(完整)G6 PAMAM 树状聚合物 Polyfect (PF) 对培养的 HEK 293 细胞中 EGFR 酪氨酸激酶信号通路(通过细胞外信号调节激酶 1/2 (ERK1/2) 和 p38 丝裂原活化蛋白激酶 (MAPK))的影响,然后比较了单次腹腔注射 PF 或 SF 对正常和糖尿病雄性 Wistar 大鼠肾脏中 EGFR 信号通路的体内影响。结果表明,Polyfect 对 HEK-293 细胞中 EGFR、ERK1/2 和 p38 MAPK 的磷酸化具有剂量和时间依赖性的抑制作用,与选择性 EGFR 抑制剂 AG1478 的作用相似。将树状聚合物注射到非糖尿病或糖尿病动物体内24小时后发现,PF抑制了两组动物肾脏中的EGFR磷酸化,而SF则刺激了EGFR磷酸化。PF介导的EGFR磷酸化抑制以及SF或PF介导的HEK 293细胞凋亡均可被抗氧化剂(如替莫唑胺)的联合治疗显著逆转,表明这两种效应均涉及氧化应激依赖性机制。这些结果首次表明,SF和PF PAMAM树状聚合物可在体内差异性地调节重要的EGFR信号转导通路,并可能代表一类新型的EGFR调节剂。这些发现可能对PAMAM树状聚合物在纳米医学中的应用具有重要的临床意义。[3]综上所述,我们在此鉴定出PI4KA是酪氨酸激酶抑制剂AG1478的一个新的细胞靶点。AG1478此前仅被认为是一种EGFR抑制剂和高尔基体分散剂。我们发现AG1478对EMCV和HCV具有抗病毒活性,并证实其作用机制涉及抑制PI4KA活性。我们的体外实验数据表明AG1478是PI4KA的直接抑制剂;然而,我们不能排除AG1478间接或通过两种途径共同作用于PI4KA活性的可能性。AG1478的抗病毒活性很可能与其对EGFR信号通路的影响无关,因为AG1478在低纳摩尔浓度范围内即可抑制EGFR(31),而抑制病毒复制(以及PI4KA活性)则需要微摩尔浓度。尽管EGFR抑制不太可能是AG1478抗病毒活性的原因,但未来研究AL-9(以及其他结构相关的抑制剂)是否也具有抗EGFR特性将很有意义。总之,我们的研究揭示了酪氨酸激酶抑制剂AG1478的重要细胞效应和抗病毒特性,该化合物此前被认为是一种有前景的癌症化疗药物。[4]
|
| 分子式 |
C16H15CL2N3O2
|
|
|---|---|---|
| 分子量 |
352.21
|
|
| 精确质量 |
351.054
|
|
| 元素分析 |
C, 54.56; H, 4.29; Cl, 20.13; N, 11.93; O, 9.08
|
|
| CAS号 |
170449-18-0
|
|
| 相关CAS号 |
AG-1478;153436-53-4
|
|
| PubChem CID |
3035187
|
|
| 外观&性状 |
Typically exists as solid at room temperature
|
|
| 密度 |
1.337g/cm3
|
|
| 沸点 |
458.5ºC at 760mmHg
|
|
| 闪点 |
231.1ºC
|
|
| 蒸汽压 |
1.37E-08mmHg at 25°C
|
|
| LogP |
4.919
|
|
| tPSA |
56.27
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
5
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
23
|
|
| 分子复杂度/Complexity |
360
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
ClC1=C([H])C([H])=C([H])C(=C1[H])N([H])C1C2=C([H])C(=C(C([H])=C2N=C([H])N=1)OC([H])([H])[H])OC([H])([H])[H].Cl[H]
|
|
| InChi Key |
WDJDYIUSDDVWKB-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C16H14ClN3O2.ClH/c1-21-14-7-12-13(8-15(14)22-2)18-9-19-16(12)20-11-5-3-4-10(17)6-11;/h3-9H,1-2H3,(H,18,19,20);1H
|
|
| 化学名 |
N-(3-chlorophenyl)-6,7-dimethoxyquinazolin-4-amine;hydrochloride
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8392 mL | 14.1961 mL | 28.3922 mL | |
| 5 mM | 0.5678 mL | 2.8392 mL | 5.6784 mL | |
| 10 mM | 0.2839 mL | 1.4196 mL | 2.8392 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Small molecule EGFR inhibitors attenuate HFD-induced EGFR phosphorylation and myocardial fibrosis in ApoE−/−mouse hearts.Sci Rep.2016 Apr 18;6:24580. |
|---|
 542 or AG1478 suppress HFD-induced inflammation in ApoE−/−mouse hearts.Sci Rep.2016 Apr 18;6:24580. |
 EGFR inhibitors reverse HFD-induced hypertrophic remodeling, fibrosis and apoptosis in C57BL/6 mouse heart.Sci Rep.2016 Apr 18;6:24580. |
 EGFR inhibitors attenuate PA-induced inflammation in H9C2 Cells.Sci Rep.2016 Apr 18;6:24580. |
|---|
 EGFR inhibitors reverse PA-induced hypertrophy, fibrosis and apoptosis in H9C2 cells.Sci Rep.2016 Apr 18;6:24580. |
 PA induces EGFR activation through TLR4/c-Src signaling cascade in H9C2 cells.Sci Rep.2016 Apr 18;6:24580. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
