| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
Human soluble adenylyl cyclase (sAC, AC10) - inhibitor (IC50 = 4.0 ± 0.2 μM) [2]
|
|---|---|
| 体外研究 (In Vitro) |
在盐水中,联硫醇(0.1–10 mg/mL,72 小时)对新鲜的副阿米巴寄生虫表现出毒性 [1]。腺苷酸环化酶 (AC) 活性被联硫醇 (0-100 μM) 抑制,达到 IC50 值。 Bithionol(50 和 100 μM,0-10 分钟)可降低 cAMP 并几乎抑制 sAC。在过度表达 4-4 累积影响的细胞中,sAC 增强 cAMP。
酶抑制活性:在含有5 mM ATP、10 mM MgCl2和10 mM CaCl2的测定条件下,对纯化的人可溶性腺苷酸环化酶催化域 (sAC-cat,残基1-469) 进行的剂量反应实验表明,Bithionol能有效抑制sAC活性,IC50为4.0 ± 0.2 μM。其效力与已知的sAC抑制剂KH7及其类似物六氯酚 (IC50 = 1.6 ± 0.1 μM) 相当。[2] 结合亲和力:微量热泳动测量确定了Bithionol与apo-sAC蛋白的结合亲和力 (KD) 为0.43 ± 0.06 μM,表明存在高亲和力相互作用。[2] 抑制机制:动力学分析(在不同抑制剂浓度下的ATP滴定)表明,Bithionol是sAC的混合型抑制剂。它导致表观Vmax显著降低,对底物Km的增效应较弱,表明其对ATP主要是非竞争性抑制,兼有较小的竞争性成分。用混合抑制模型直接拟合得到的Ki为2.3 μM。该抑制作用似乎与生理激活剂碳酸氢根存在竞争关系,因为增加碳酸氢根浓度会使IC50值升高(从无碳酸氢根时的6 μM升高到40 mM碳酸氢根时的11 μM)。[2] 抑制的结构基础(X射线晶体学):解析了人sAC催化域与Bithionol复合物的晶体结构,分辨率为2.24 Å。结构显示,Bithionol深入结合在变构碳酸氢根结合位点 (BBS) 中,并占据了连接该调节位点与活性位点的通道。关键相互作用包括与Val167、Ala100、Leu162、Leu102、Val175以及由Phe338/Phe296形成的疏水补丁的疏水接触,以及与Phe45和Phe336的T型π-π堆积。一个羟基与Met337的骨架酰胺形成氢键。氯原子可能与Lys95和骨架原子形成极性相互作用。活性位点和变构位点之间通讯的关键残基Arg176与Bithionol的π电子相互作用。结合诱导了构象变化,包括底物结合位点的收紧和催化残基Asp99的位移,这阻碍了有活性的酶-底物复合物的形成。[2] 突变分析:与野生型sAC相比,抑制sAC-R176A突变体需要2-3倍更高浓度的Bithionol,证实了Arg176参与抑制剂结合和抑制机制。[2] 对跨膜ACs (tmACs) 的选择性:在sAC敲除的小鼠胚胎成纤维细胞中(其cAMP产生完全来自tmACs),高达100 μM的Bithionol不抑制福司可林刺激的cAMP积累,而tmAC特异的P位点抑制剂2',5'-双脱氧腺苷则具有抑制作用。这证明在细胞环境中,Bithionol相对于tmACs对sAC具有特异性。[2] |
| 体内研究 (In Vivo) |
在小鼠中,联硫醇对未成熟的 H. nana 表现出中等功效(100 mg/kg/天,侧壁粉末,持续 12 天)[3]。
|
| 酶活实验 |
sAC活性测定:活性测定使用100 ng纯化的人sAC-cat蛋白,在含有50 mM Tris/HCl (pH 8.0)、50 mM NaCl、10 mM MgCl2、10 mM CaCl2和5 mM ATP的缓冲液中进行。反应在37°C下孵育,并通过快速冷冻终止。生成的cAMP使用以下三种方法之一进行定量:1) 测量³²P标记的cAMP,2) RapidFire质谱法,或 3) 在UPLC系统上使用C18柱进行反相色谱分析,等度洗脱流动相为97% 20 mM醋酸铵 (pH 4.5) 和3% 乙腈。对于抑制研究,化合物在不同浓度下测试,数据拟合到适当的抑制模型。[2]
结合实验(微量热泳动):使用微量热泳动法测量Bithionol与sAC的结合亲和力。使用0.4 μM的sAC-cat蛋白,缓冲液含有50 mM Tris/HCl (pH 8)、50 mM NaCl和15 mM CaCl2。KD值通过将结合转变曲线拟合到单位点结合方程来确定。[2] |
| 细胞实验 |
细胞cAMP积累实验(sAC过表达):为了测试Bithionol的细胞活性,在稳定过表达sAC的培养4-4细胞中测量cAMP积累。用500 μM异丁基甲基黄嘌呤抑制细胞磷酸二酯酶。与抑制剂孵育指定时间后,使用直接cAMP酶免疫分析法定量细胞cAMP。Bithionol引起cAMP形成的剂量依赖性减少,在100 μM时几乎完全抑制了sAC依赖的cAMP积累,效果与已知的sAC抑制剂KH7 (30 μM) 相似。[2]
细胞cAMP积累实验(tmAC活性):为了评估选择性,在sAC敲除的小鼠胚胎成纤维细胞中测量福司可林刺激的cAMP积累。细胞在存在或不存在抑制剂的情况下用50 μM福司可林处理。如上所述定量细胞cAMP。100 μM的Bithionol不抑制这种tmAC依赖的cAMP积累。[2] |
| 动物实验 |
动物/疾病模型:感染了未成熟矮小绦虫的小鼠[3]
剂量:100 mg/kg/天 给药途径:口服,感染后12天 实验结果:48%的成熟矮小绦虫被清除。LD50:760 mg/kg。 |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
联硫酚在宿主消化道的吸收程度有限,主要在血液中检测到,尤其是在胆汁中检测到,并最终通过胆汁排出体外。给药后2小时内,胆汁中的药物浓度达到峰值。血液中的药物浓度显著低于胆汁中的浓度。 当给大鼠喂食(35)S-联硫酚、(35)S-联硫酚亚砜或(35)S-联硫酚砜时,代谢物的尿液排泄量非常低。联硫酚亚砜主要通过粪便排出。超过90%的胆汁放射性以三种化合物的葡萄糖醛酸苷形式存在,其中超过70%的葡萄糖醛酸苷为硫双酚葡萄糖醛酸苷。 代谢/代谢物 当大鼠口服(35)S-硫双酚亚砜后,收集尿液、粪便和胆汁。在尿液中观察到八种代谢物。纸层析和化学测试鉴定出其中一种为无机硫酸盐。一种强酸被鉴定为3,5-二氯-2-羟基磺酸。另外两种化合物被鉴定为硫双酚砜和硫双酚。此外,还鉴定出三种化合物为硫双酚、硫双酚亚砜和硫双酚砜的葡萄糖醛酸苷。还有一种代谢物未被鉴定。除游离的硫双酚砜外,胆汁中发现的代谢物与尿液中发现的代谢物相同。然而,定量差异很大。硫双酚砜的葡萄糖醛酸苷占胆汁中 (35)S 的 71%,但在尿液中仅占 16.5%。/硫双酚亚砜/ 当给大鼠喂食 (35)S-硫双酚、(35)S-硫双酚亚砜或 (35)S-硫双酚砜时,代谢物的尿排泄量……非常低。……超过 90% 的胆汁放射性以这 3 种化合物的葡萄糖醛酸苷的形式存在,其中超过 70% 的葡萄糖醛酸苷以硫双酚葡萄糖醛酸苷的形式存在。砜代谢生成儿茶酚和愈创木酚。 /硫双酚和硫双酚砜是硫双酚亚砜的主要代谢产物。/ |
| 毒性/毒理 (Toxicokinetics/TK) |
相互作用
当比硫酚与四氯化碳、酒石酸锑钠、盐酸依米丁、六氯乙烷或六氯对二甲苯联合使用时,毒性会中度至显著增强。 非人类毒性值 大鼠口服LD50:1430 mg/kg 小鼠口服LD50:2100 mg/kg 小鼠静脉注射LD50:18 mg/kg 一般细胞毒性:作者指出,他们观察到比硫酚在浓度约为10 μM时开始出现一般细胞毒性作用,该浓度是细胞环境中显著抑制sAC所需的浓度。 [2] 历史上的FDA禁令:比硫酚曾被用作化妆品中的抗菌剂,但由于其光敏性,美国食品药品监督管理局(FDA)禁止了这种用途。[2] |
| 参考文献 |
|
| 其他信息 |
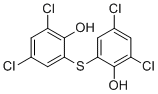
2,2'-硫代双(4,6-二氯苯酚)为白色或灰白色结晶性粉末,具有非常淡的芳香或酚类气味。(NTP, 1992)
硫代双酚是一种芳基硫醚,属于二苯基硫醚,其中每个苯基的2位被羟基取代,3位和5位被氯取代。它是一种杀真菌剂和驱虫剂,曾用于多种外用药物中治疗肝吸虫病,但由于被证实是一种强效光敏剂,可能引起严重的皮肤疾病,因此被撤市。它目前可用作抗扁形动物药物和抗真菌农药。它是一种芳基硫醚、有机氯杀虫剂、二氯苯、多酚、桥联二苯基杀菌剂和桥联二苯基抗真菌药物。 比硫酚曾作为多种外用药物的活性成分上市,但已被证实是一种强效光敏剂,可能导致严重的皮肤疾病。1967年10月24日,比硫酚药物产品的新药申请被撤销(参见1967年10月31日《联邦公报》(32 FR 15046))。 一种用于治疗吸虫和绦虫感染的卤代抗感染剂。 作用机制 比硫酚干扰蠕虫(目标物种)的神经肌肉生理,损害其卵的形成,并可能导致覆盖蠕虫的保护性角质层出现缺陷。在生化水平上,氧化磷酸化受到抑制,并且硫双酚分子可以螯合铁,从而使含铁酶系统失活。在目标物种中,体内用硫双酚治疗成虫可降低糖酵解和氧化代谢。具体而言,琥珀酸氧化受到抑制。虽然硫双酚的确切作用机制尚不清楚,但推测其作用机制可能依赖于酚羟基作为氢受体,这些氢原本会参与与琥珀酸氧化相关的反应。干扰这些反应或许会使吸虫缺乏维持生命所需的能量。 治疗用途 局部抗感染剂;抗扁形动物药物 硫双酚用于治疗肺吸虫病,剂量为30至50 mg/kg体重,隔日口服,共10至15次。在治疗肝吸虫病时,也使用了相同的剂量。在治疗肝片吸虫病时,也曾使用过硫双酚,剂量最高可达每日3克,隔日服用,共15次。在绦虫感染中,曾使用过最高剂量为60毫克/公斤体重的硫双酚,分两次服用,间隔约1小时。 肺部感染患者中,0.8%会发生脑部感染。在一组24例脑部感染患者中,硫双酚对所有患者均有效,能够清除痰液中的虫卵并终止铁锈痰的产生。然而,仅有9例患者服用该药后脑部症状得到有效控制,包括视力丧失、明显的脑膜炎以及1例硬膜内脓肿。 该药需与食物同服,以降低胃肠道症状的发生率和严重程度。 有关BITHIONOL(共16种)的更多治疗用途(完整)数据,请访问HSDB记录页面。 背景和治疗潜力:可溶性腺苷酸环化酶(sAC)是糖尿病、青光眼等疾病的潜在治疗靶点,也可作为男性避孕药。Bithionol是一种新型sAC抑制剂,它通过与生理激活剂(碳酸氢盐)结合的变构机制发挥作用。[2] 作用机制:Bithionol是一种强效的sAC特异性抑制剂,其作用机制主要为非竞争性变构。它与sAC独特的碳酸氢根结合位点(BBS)结合,诱导构象变化,从而破坏活性位点并阻碍催化。这是首个已知通过这种机制作用于该位点的sAC抑制剂。[2] 化学骨架和药物开发潜力:尽管比硫酚及其类似物六氯酚在有效浓度下具有多效性生物学效应(抗病毒、抗菌、抑制癌细胞生长)和细胞毒性,使其本身不适合作为药物,但它们的化学骨架、结合位点和抑制机制为开发更具特异性和更强效的sAC靶向治疗药物提供了宝贵的起点。[2] 与其他抑制剂的比较:比硫酚与BBS的结合深度比弱抑制剂DIDS更深。与之前描述的强效抑制剂 ASI-8(其作用范围从 BBS 延伸至底物结合位点)不同,Bithionol 主要占据调节位点,从而导致其独特的、主要为变构抑制的特性。[2] |
| 分子式 |
C12H6CL4O2S
|
|---|---|
| 分子量 |
356.038
|
| 精确质量 |
353.884
|
| CAS号 |
97-18-7
|
| 相关CAS号 |
Bithionol (sulfoxide);844-26-8
|
| PubChem CID |
2406
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.8±0.1 g/cm3
|
| 沸点 |
444.7±45.0 °C at 760 mmHg
|
| 熔点 |
188°C
|
| 闪点 |
222.8±28.7 °C
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
| 折射率 |
1.741
|
| LogP |
5.51
|
| tPSA |
65.76
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
19
|
| 分子复杂度/Complexity |
282
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
JFIOVJDNOJYLKP-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C12H6Cl4O2S/c13-5-1-7(15)11(17)9(3-5)19-10-4-6(14)2-8(16)12(10)18/h1-4,17-18H
|
| 化学名 |
2,4-dichloro-6-(3,5-dichloro-2-hydroxyphenyl)sulfanylphenol
|
| 别名 |
Bithionol CP 3438 Bitin CP3438 Lorothidol CP-3438
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ≥ 33 mg/mL (~92.68 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (5.84 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (5.84 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8087 mL | 14.0434 mL | 28.0867 mL | |
| 5 mM | 0.5617 mL | 2.8087 mL | 5.6173 mL | |
| 10 mM | 0.2809 mL | 1.4043 mL | 2.8087 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1



































 463611831
463611831
