| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
Camptothecins; TOP I; topoisomerase I
|
|---|---|
| 体外研究 (In Vitro) |
抗体-药物偶联物可选择性、高效地将抗癌药物递送至肿瘤组织,具有显着的抗肿瘤功效和广泛的治疗窗[2]。使用具有不同生物学特性的五种 CRC 细胞系研究了 [fam-] trastuzumab deruxtecan 对 CRC 的抗肿瘤活性。首先检查了这些不同细胞系中 HER2 在 mRNA 和蛋白质水平上的表达。免疫印迹分析以及 RT 和实时聚合酶链反应 (PCR) 分析显示,所有 CRC 细胞系中 HER2 蛋白和 HER2 mRNA 的量均远小于 NCI-N87 细胞。 [fam-] trastuzumab deruxtecan 减弱了 NCI-N87 细胞的活力,与之前的结果一致,而所有五种 CRC 细胞系均表现出对该药物的耐药性。这些发现表明 HER2 蛋白的表达水平可能决定对 [fam-] 曲妥珠单抗 deruxtecan 的敏感性。
Dxd(ADC的Exatecan衍生物)是一种强效的DNA拓扑异构酶I抑制剂,用作与HER2靶向ADC的偶联药物(DS-8201a),IC50为0.31μM。IC50范围为1.43 nM至4.07 nM,Dxd对KPL-4、NCI-N87、SK-BR-3和MDA-MB-468的人癌症细胞系具有细胞毒性;然而,以Dxd为有效载荷的对照IgG-ADC对四种细胞系(表达HER2)没有抑制作用。IC50值分别为26.8、25.4和6.7 ng/mL,DS-8201a(有效载荷为Dxd)对HER2阳性KPL-4、NCI-N87和SK-BR-3细胞系表现出显著的抑制作用,但对MDA-MB-468没有这种抑制作用(IC50,>10000 ng/mL)[3]。 DS-8201a的结构如图1A所示DS-8201a是一种靶向HER2的ADC,由抗HER2抗体和拓扑异构酶I抑制剂DX-8951(Dxd)的衍生物组成,它们通过马来酰亚胺-甘油-苯丙氨酸-甘油(GGFG)肽接头结合在一起。在用还原剂三(2-羧乙基)膦盐酸盐(TCEP HCl)还原链间二硫键后,接头有效载荷通过半胱氨酸残基与抗体偶联。由于四肽被溶酶体酶(如组织蛋白酶B和L)分解,这些酶在肿瘤细胞中高度表达,因此假设DS-8201a被溶酶体酶切割并释放Dxd,Dxd在与HER2受体结合后特异性攻击肿瘤细胞中的靶分子,并在肿瘤细胞内内化。通过使用RPC,DS-8201a的DAR被确定为约8,这是传统链间半胱氨酸偶联的理论最大载药量。因此,在HIC图中观察到均匀的药物分布(图1B)。我们证实,Dxd在拓扑异构酶I的抑制活性方面比SN-38和DX-8951f更有效,这是通过拓扑异酶I介导的DNA松弛试验测量的(图1C)[3]。 DS-8201a[3] 对癌症细胞生长的抑制作用 将DS-8201a对癌症细胞生长的抑制活性与体外与Dxd结合的抗HER2 Ab和对照IgG–ADC对各种人类癌症细胞系的抑制活性进行比较。首先通过流式细胞术分析评估细胞系KPL-4、NCI-N87、SK-BR-3和MDA-MB-468细胞表面HER2的表达(图2A)。KPL-4、NCI-N87和SK-BR-3的相对MFI分别为95.7、101.6和56.2,表明HER2在细胞表面上明确表达,而MDA-MB-468的相对MFI为1.0,表明MDA-MB-465中没有表达。观察到DS-8201a对HER2阳性KPL-4、NCI-N87和SK-BR-3具有显著的细胞生长抑制活性,IC50值分别为26.8、25.4和6.7 ng/mL,而对MDA-MB-468没有这种抑制作用,IC50值>10000 ng/mL(图2B)。尽管抗HER2抗体显示出对NCI-N87和SK-BR-3的细胞生长抑制活性,但这些活性比DS-8201a弱得多;NCI-N87和SK-BR-3的IC50值分别为204.2和65.9 ng/mL(图2B)。此外,对照IgG ADC在四种细胞系中均未显示出细胞生长抑制活性(图2B),尽管所有四种细胞株对有效载荷Dxd都很敏感(IC50:1.43 nmol/L-4.07 nmol/L)。这些结果表明,药物与抗HER2抗体结合显著增强了DS-8201a的细胞生长抑制活性,并且DS-8201a对HER2阳性细胞系显示出靶向特异性生长抑制作用。 |
| 体内研究 (In Vivo) |
测试了 [fam-] 曲妥珠单抗 deruxtecan 在表达 HER2 的异种移植肿瘤模型中的疗效。首次通过免疫组化(IHC)证实HCT116-Mock、HCT116-H2L或HCT116-H2H细胞裸鼠皮下肿瘤中HER2蛋白表达水平。以3.0 mg/kg剂量施用[fam-]曲妥珠单抗deruxtecan显着抑制由HCT116-H2L或HCT116-H2H细胞形成的肿瘤的生长,但不抑制由HCT116-Mock细胞形成的肿瘤的生长。第 24 天时,与 PBS 载体相比,[fam-] trastuzumab deruxtecan 对 HCT116-H2L 和 HCT116-H2H 细胞的抑制程度分别为 60% 和 93%。[fam-] trastuzumab deruxtecan 治疗对身体没有影响三组小鼠中任意一组的体重。因此,这些发现表明,异种移植模型中肿瘤对[fam-]曲妥珠单抗deruxtecan的敏感性取决于HER2表达水平,并且这种治疗与明显的毒性无关。
DS-8201a(DXdis为有效载荷,10mg/kg,静脉注射)在HER2 IHC 1+/FISH阴性表达的HER2低表达ST565和ST313模型以及KPL4、JIMT-1和Capan-1的HER2阳性模型中显示出强大的抗肿瘤活性[3]。 体内抗肿瘤活性[3] 在HER2阳性NCI-N87异种移植物模型中评估了DS-8201a的体内抗肿瘤活性DS-8201a以剂量依赖的方式诱导肿瘤生长抑制,单次剂量超过1mg/kg时肿瘤消退,而不会诱导小鼠的一般状况或体重变化出现任何异常(图2C)。在同一模型中,4 mg/kg的抗HER2抗体给药部分抑制了肿瘤生长,表明在第21天与对照组相比,肿瘤生长抑制率(TGI)为31%(图2D)。另一方面,DS-8201a明显显示出更有效的抗肿瘤疗效,表明在相同的4 mg/kg剂量下,TGI为99%,因此在体内和体外模型中都观察到药物偶联的疗效增强(图2D)。此外,DS-8201a的体内疗效取决于其HER2结合,因为对照IgG-ADC没有抑制肿瘤生长(图2D)。 DS-8201a在低HER2表达肿瘤中的抗肿瘤活性[3] T-DM1已被批准用于HER2阳性转移性癌症患者,根据当前指南,其定义为HER2 IHC 3+或IHC 2+/FISH阳性,并且在FISH阴性、HER2 1+和2+人群中,HER2靶向治疗的临床需求仍有待满足。因此,在具有不同HER2表达水平的各种小鼠异种移植物模型中评估了DS-8201a的抗肿瘤活性;KPL-4(强阳性)、JIMT-1(中度阳性)、Capan-1(弱阳性)和GCIY(阴性)(图4A和B)。还评估了具有与DS-8201a相同的药物接头和约一半DAR(DAR 3.4)的抗-HER2 ADC,以研究DAR对抗肿瘤活性的影响。虽然T-DM1仅对KPL4模型有效,但DS-8201a对所有具有KPL4、JIMT-1和Capan-1的HER2阳性模型有效。两种ADC在GCIY模型中均无效。抗HER2 ADC(DAR 3.4)抑制了所有HER2阳性模型的肿瘤生长,其疗效是HER2表达依赖性的。在HER2弱阳性Capan-1模型中,DS-8201a的疗效明显强于抗HER2 ADC(DAR 3.4)。这些结果表明,高DAR ADC DS-8201a能够将足够的有效载荷量递送到癌症细胞中,表明即使在低HER2水平下也具有细胞毒性。在HER2强阳性模型的情况下,即使是低DAR ADC也能够为细胞死亡提供足够的有效载荷。DS-8201a对HER2水平较高的肿瘤有效,因为其DAR约为8。为了在HER2低表达模型中确认DS-8201a的HER2特异性,在HER2高CFPAC-1模型中进行了竞争性抑制研究(图4C)。DS-8201a的疗效被抗HER2抗体的先前治疗所抵消,对照IgG ADC在比DS-8201a高3倍的剂量下没有抑制肿瘤生长。从这些结果中,证实了DS-8201a在HER2低表达模型中的HER2特异性。 与PDX模型中的T-DM1的比较[3] 除了基于细胞系的异种移植物模型外,还进行了几次PDX评估,以更精确地评估临床效益。在癌症PDX模型中,NIBIO G016,DS-8201a表现出强大的抗肿瘤活性和肿瘤消退,但T-DM1没有(图5A)。由于该模型中的HER2状态为IHC 3+/FISH+,因此认为DS-8201a和T-DM1之间抗肿瘤疗效的差异是基于有效载荷的不同敏感性,这是由于每种有效载荷的作用机制不同。在癌症PDX模型中,尽管DS-8201a和T-DM1在HER2 IHC 2+/FISH阳性ST225模型中均有效,但在第21天,用DS-8201a而非T-DM1治疗的5只小鼠中有3只观察到肿瘤完全消退(图5B)。此外,DS-8201a在HER2 IHC 1+/FISH阴性表达的HER2低表达ST565和ST313模型中显示出抗肿瘤活性(图5C和D),但T-DM1没有。这一结果表明了与基于细胞系的异种移植物模型(如Capan-1和CFPAC-1)类似的趋势(图4B和C)。因此,在具有几种HER2表达水平的所有4种模型中,DS-8201a显示出比T-DM1更有效的抗肿瘤活性。这些结果表明,DS-8201a与T-DM1具有可区分的潜力,T-DM1在T-DM1不敏感和HER2低表达的肿瘤中显示出有效性,这是由于结合药物的不同作用机制和DS-8201a的高DAR造成的。 |
| 酶活实验 |
使用Freedom EVO200系统进行平行人工膜渗透性测定(PAMPA)。受体板的滤膜涂有GIT‐0脂质溶液。将DMSO(10mM)中的每种化合物溶液加入Prisma HT缓冲液中,获得5μM供体溶液(含有0.05%DMSO,pH 5.0和pH 7.4),然后放置在供体板上。受体板填充有受体沉缓冲液。将供体板堆叠在受体板上,并在25°C下孵育4小时。培养后,通过LC-MS/MS系统(API 4000)测量两个平板中的化合物浓度。使用PAMPA Evolution DP软件计算渗透系数(Peff;10−6 cm/s)。[1]
拓扑异构酶I抑制试验[3] SN-38、DX-8951f和DX-8951衍生物(DXd)是在内部合成的。根据之前的报告,SN-38、DX-8951f和DXd对人拓扑异构酶I的抑制活性是通过拓扑异酶I介导的DNA弛豫试验进行评估的。简而言之,重组人拓扑异构酶I与每种药物一起孵育5分钟。然后,加入超螺旋DNA pBR322,在25°C下孵育60分钟。在琼脂糖凝胶上电泳混合物后,用CCD成像仪测量超螺旋DNA的量。 DS-8201a在血浆中的体外稳定性[3] 在小鼠、大鼠、猴子和人血浆中评估了37°C下浓度为10μg/mL的DS-8201a>在21天内释放DXd的速率。 |
| 细胞实验 |
将细胞接种在96孔板中,KPL‐4为1000个细胞/孔,MDA‐MB‐468为2000个细胞/孔。培养过夜后,加入每个ADC的连续稀释溶液。5天后,根据制造商的说明,使用来自Promega的CellTiter‐Glo发光细胞活力测定法评估细胞活力。在共培养研究中,将KPL‐4和MDA‐MB‐468细胞分别以1×105细胞和3×105细胞接种在6孔板中的2 mL/孔培养基中。孵育过夜后,从板中取出上清液,并以6mL/孔的速度加入每种ADC稀释剂(10nM)。培养5天后,从平板上分离活细胞,并使用细胞计数器测定每个孔中的细胞数。为了确定KPL‐4和MDA‐MD‐468细胞在总活细胞中的比例,用抗HER2/nue-FITC对细胞进行染色,并在冰上孵育20分钟。洗涤后,使用流式细胞仪测量2×104染色细胞的荧光信号。根据每个处理井中HER2阳性和HER2阴性细胞的数量和比例,计算KPL‐4或MDA‐MB‐468细胞的数量。[1]
将细胞接种在96孔板中,KPL-4为1000个细胞/孔,MDA-MB-468为2000个细胞/孔。孵育过夜后,加入每种ADC的连续稀释溶液。5天后,根据制造商的说明,使用Promega的CellTiter̴Glo发光细胞存活率测定法评估细胞存活率。在共培养研究中,将KPL-4和MDA-MB-468细胞分别以1×105个细胞和3×105个电池接种在2 mL/孔培养基中的6孔板中。孵育过夜后,从培养板中取出上清液,以6mL/孔的速度加入每种ADC稀释剂(10nM)。培养5天后,从培养板上分离活细胞,并使用细胞计数器测定每个孔中的细胞数量。为了确定KPL-4和MDA-MD-468细胞在总活细胞中的比例,用抗HER2/nue FITC对细胞进行染色,并在冰上孵育20分钟。洗涤后,使用流式细胞仪测量2×104个染色细胞的荧光信号。根据每个处理孔中HER2阳性和HER2阴性细胞的数量和比例,计算KPL-4或MDA-MB-468细胞的数量。[1] 96孔板每孔接种1000个细胞。在孵育一晚后加入DXd。CellTiter Glo发光细胞活力测定用于在六天后评估细胞活力。为了鉴定每个细胞系中的HER2表达,将FITC小鼠IgG1、κ同种型对照或抗HER2/neu FITC在冰上孵育30分钟。洗涤后,使用FACSCalibur分析标记的细胞。相对平均荧光强度(rMFI)的计算已完成[3]。 细胞毒性试验[3] 将细胞以每孔1000个细胞的速度接种到96孔板上。孵育过夜后,加入每种稀释物质。根据制造商的说明,在6天后使用CellTiter Glo发光细胞存活率测定法评估细胞存活率。为了检测每个细胞系中的HER2表达,将细胞与FITC小鼠IgG1、κ同种型对照或抗HER2/neu FITC在冰上孵育30分钟,用FACSCalibur分析标记的细胞。相对平均荧光强度(rMFI)通过以下方程式计算: ELISA[3] 对于结合试验,免疫板在包被缓冲液中用2.5μg/mL His标记的HER2-ECD蛋白包被,并在4°C下保持过夜。洗涤后,将板封闭,并将每种连续稀释的物质加入孔中。在37°C下孵育1.5小时后,清洗平板,并在37°C下用HRP偶联的抗人IgG二抗孵育1小时。洗涤后,加入TMB溶液,用微孔板读数器测量每个孔中的A450。为了检测磷酸化Akt(pAkt),将SK-BR-3细胞在96孔板中预孵育4天,然后与每种物质孵育24小时。孵育后,根据制造商的说明,裂解细胞,并使用PathScan Phospho-Akt1(Ser473)夹心ELISA试剂盒和PathScan total-Akt1夹心ELISA试剂袋检测细胞间pAkt和总Akt。通过将处理过的归一化pAkt值除以未处理的归一化pAk值来计算每个样品孔的相对pAkt。 ADCC评估[3] 使用来源于供体的人外周血单核细胞(PBMC)作为效应细胞,SK-BR-3细胞作为靶细胞,评估抗体依赖性细胞介导的细胞毒性(ADCC)活性。效应细胞(2×105个细胞)和51Cr标记的靶细胞(1×104个细胞)与每种物质一起孵育,指示效应:靶(E:T)比为20:1。孵育4小时后,通过培养上清液中的放射性测量ADCC活性。 免疫印迹[3] 用每种物质处理KPL-4细胞。24、48或72小时后,收获细胞并用含有Halt蛋白酶和磷酸酶抑制剂混合物的M-PER裂解缓冲液裂解。 通过SDS-PAGE对样品进行装载和分离,并将其印迹到聚偏二氟乙烯膜上。将膜封闭,并用抗磷酸化Chk1(Ser345;133D3)兔单克隆抗体、抗Chk1(2G1D5)小鼠单克隆抗体、反切割PARP(Asp214)抗体、抗β-肌动蛋白(8H10D10)小鼠mAb、抗磷酸化组蛋白H2A探测过夜。X(Ser139)抗体和抗组蛋白H2A。4°C下的X抗体。然后,用SNAP皮内洗涤膜并用荧光标记的二抗孵育10分钟。使用Odyssey成像系统检测荧光信号。 |
| 动物实验 |
体内异种移植研究
所有体内研究均按照机构动物护理和使用委员会的当地指南进行。使用5周龄的特定病原体清除级雌性CAnN.Cg-Foxn1nu/CrlCrlj小鼠(BALB/c裸鼠)。所有模型均通过皮下注射接种于小鼠侧腹建立。NCI-N87和MDA-MB-468-Luc模型分别通过注射悬浮于Matrigel基质中的5 × 10⁶和1 × 10⁷个细胞建立。NCI-N87模型接种6天后,MDA-MB-468-Luc模型接种9天后,根据肿瘤体积将荷瘤小鼠随机分为治疗组和对照组,并开始给药(第0天)。 每种ADC均以3或10 mg/kg的剂量,10 mL/kg的体积,静脉注射给小鼠。作为载体,使用与ADC相同体积的ABS缓冲液(10 mM醋酸盐缓冲液,5%山梨醇,pH 5.5)。肿瘤体积定义为1/2 × 长 × 宽²。[1] 体内荧光素酶成像 将5 × 10⁶个NCI-N87细胞和1 × 10⁷个MDA-MB-468-Luc细胞悬浮于Matrigel基质中,接种到小鼠右侧腹部,总体积为100 μL。接种7天后,根据肿瘤体积将荷瘤小鼠随机分为治疗组和对照组,并开始给药(第0天)。每种ADC或载体均静脉注射给小鼠。每只小鼠的荧光素酶活性均使用体内成像系统每周测量两次,同时在静脉注射150 mg/kg荧光素10分钟后测量肿瘤长度。发光量使用分析软件分析,结果以平均辐射强度(p/s/cm²/sr)表示。在另一项研究中,除将混合物接种到小鼠右侧腹部外,还将密度为1.5 × 10⁷个细胞的MDA-MB-468-Luc细胞接种到小鼠左侧腹部。接种7天后,以与上述类似的方式进行给药和评估。[1] 小鼠:简而言之,将每种细胞悬液或肿瘤碎片皮下注射到特定病原体清除(SPF)的雌性裸鼠中。从第0天开始给药,待肿瘤生长到合适大小后,根据肿瘤体积将荷瘤小鼠随机分为治疗组和对照组。小鼠接受静脉注射DS-8201a(1或10 mg/kg,静脉注射;Dxd为有效载荷)。计算肿瘤生长抑制率(TGI,%)[1]。 细胞系和患者来源的异种移植研究[3] 详细的研究步骤见补充材料。简而言之,将每种细胞悬液或肿瘤碎片皮下接种到特定病原体清除(SPF)雌性裸鼠体内。当肿瘤生长到适当体积后,根据肿瘤体积将荷瘤小鼠随机分为治疗组和对照组,并于第0天开始给药。每种物质均通过静脉注射给药。肿瘤生长抑制率(TGI,%)根据以下公式计算: 食蟹猴体内DS-8201a的药代动力学[3] 采用经验证的配体结合试验测定血浆中DS-8201a和总抗体的浓度;定量下限为0.100 μg/mL。采用经验证的液相色谱-串联质谱(LC/MS-MS)法测定血浆中DXd的浓度;定量下限为0.100 ng/mL。DS-8201a以3.0 mg/kg的剂量静脉注射给雄性食蟹猴。给药后 672 小时内测量血浆中 DS-8201a、总抗体和 DXd 的浓度。 |
| 药代性质 (ADME/PK) |
食蟹猴体内的药代动力学[3]
单次静脉注射DS-8201a后,血浆中DS-8201a浓度呈指数下降。DS-8201a和总抗体的稳态分布容积(Vss)接近血浆容积(数据未显示)。DS-8201a和总抗体的药代动力学特征未见明显差异,表明DS-8201a的肽连接子即使在DAR 8时在血浆中也保持稳定(图2E)。仅在有限的时间点检测到低水平的DXd(图2E)。 体外血浆稳定性[3] 在小鼠、大鼠、猴和人血浆中,第21天DXd从DS-8201a中的释放率在1.2%至3.9%之间(图2F),与其他抗体偶联药物(ADC)如T-DM1、SGN-35(Brentuximab vedotin)和inotuzumab ozogamicin的释放率相当或更低(35-37)。这些结果表明DS-8201a在血浆中稳定。 |
| 毒性/毒理 (Toxicokinetics/TK) |
DS-8201a 的安全性概况 [3]
在食蟹猴(DS-8201a 的交叉反应物种)和大鼠(抗原非结合物种;表 1)中进行了重复静脉给药(每 3 周一次,共 3 次)研究。在大鼠研究中,剂量高达 197 mg/kg(最大剂量)时,未发现死亡或危及生命的毒性。因此,动物严重毒性剂量 10% (STD10) 被认为 >197 mg/kg。在猴子研究中,一只雌性猴子在最高剂量 78.8 mg/kg 下于第 26 天因濒死而被实施安乐死。濒死的原因似乎是该动物状况恶化,包括体重和食物摄入量下降,以及骨髓毒性和肠道毒性。补充表S1显示了存活猴子肠道、骨髓和肺脏的显微镜检查结果。胃肠道毒性和骨髓毒性是拓扑异构酶I抑制剂临床应用中典型的剂量限制因素。DS-8201a对肠道的影响非常轻微,在剂量高达78.8 mg/kg时,所有动物均未出现明显的肠道病变。骨髓毒性仅在78.8 mg/kg剂量下出现,并伴有网织红细胞比例下降。在10 mg/kg和30 mg/kg剂量下,猴子的白细胞和红细胞计数均未见异常。重复给予猴子 DS-8201a 78.8 mg/kg 剂量后,引起中度肺毒性;在 ≥30 mg/kg 剂量下,经过 6 周恢复期后,毒性分级为轻度或极轻度。基于上述死亡率和毒性严重程度,猴子的最高非严重毒性剂量 (HNSTD) 被确定为 30 mg/kg。在与临床方案相对应的重复给药后,大鼠和猴子在剂量高达 197 mg/kg 和 30 mg/kg 时均耐受性良好,且非临床安全性数据符合人体试验的要求。 |
| 参考文献 | |
| 其他信息 |
抗体药物偶联物能够选择性且高效地将抗癌药物递送至肿瘤组织,并具有显著的抗肿瘤疗效和较宽的治疗窗口。DS-8201a 是一种靶向人表皮生长因子受体 2 (HER2) 的抗体药物偶联物,采用新型连接子-有效载荷系统,并结合强效拓扑异构酶 I 抑制剂依沙替康衍生物(DX-8951 衍生物,DXd)制备而成。它对曲妥珠单抗-美坦新 (T-DM1) 不敏感的患者来源异种移植瘤模型(无论 HER2 高表达还是低表达)均有效。本研究评估了 DS-8201a 的旁观者杀伤效应,并与 T-DM1 进行了比较。我们证实,DS-8201a 的有效载荷 DXd (1) 具有高膜渗透性,而 T-DM1 的有效载荷 Lys-SMCC-DM1 的膜渗透性较低。在体外HER2阳性KPL-4细胞和阴性MDA-MB-468细胞的共培养条件下,DS-8201a可杀死两种细胞,而T-DM1和低渗透性有效载荷的抗体药物偶联物anti-HER2-DXd(2)则无此作用。体内评估采用体内成像系统,将小鼠接种HER2阳性NCI-N87细胞和HER2阴性MDA-MB-468-Luc细胞的混合物。体内实验表明,DS-8201a可降低小鼠的荧光素酶信号,表明其抑制了MDA-MB-468-Luc细胞群;然而,T-DM1和anti-HER2-DXd(2)则无此作用。此外,证实DS-8201a对接种在NCI-N87肿瘤对面的MDA-MB-468-Luc肿瘤无效,这表明DS-8201a的旁观者杀伤效应仅在HER2阳性细胞附近的细胞中观察到,表明全身毒性方面无需过度担忧。这些结果表明,DS-8201a由于其高膜渗透性的有效载荷而具有强大的旁观者效应,并且对治疗对T-DM1无反应的HER2异质性肿瘤有益。[1]
目的:开发了一种新型抗肿瘤候选药物——含有新型拓扑异构酶I抑制剂的抗HER2抗体药物偶联物DS-8201a,并对其临床前药理学特性进行了评估。 实验设计:在多种HER2阳性细胞系和患者来源的异种移植(PDX)模型中,评估了DS-8201a的体外和体内药理活性,并与T-DM1进行了比较。同时,还评估了其疗效的作用机制。本研究评估了DS-8201a在食蟹猴体内的药代动力学以及在大鼠和食蟹猴体内的安全性。 结果:DS-8201a表现出HER2表达依赖性的细胞生长抑制活性,并在HER2阳性胃癌NCI-N87模型中,单次给药剂量超过1 mg/kg即可诱导肿瘤消退。DS-8201a与HER2的结合活性和ADCC活性与未偶联的抗HER2抗体相当。DS-8201a还显示出对Akt磷酸化的抑制活性。DS-8201a可诱导Chk1和组蛋白H2A.X(DNA损伤标志物)的磷酸化。 DS-8201a 的药代动力学和安全性良好,在食蟹猴中,最高非严重毒性剂量为 30 mg/kg,表明 DS-8201a 在人体中具有良好的耐受性。DS-8201a 在 T-DM1 不敏感且 HER2 高表达的 PDX 模型中有效。DS-8201a(而非 T-DM1)对多种 HER2 低表达的乳腺癌 PDX 模型显示出抗肿瘤疗效。结论:DS-8201a 在多种 HER2 阳性模型中表现出强效的抗肿瘤活性,且具有良好的药代动力学和安全性。结果表明,DS-8201a 将成为一种有价值的疗法,具有治疗 T-DM1 不敏感的 HER2 阳性癌症和 HER2 低表达癌症的巨大潜力。Clin Cancer Res; 22(20); 5097-108. ©2016 AACR. [3] 目前市售和临床开发中的大多数抗体偶联药物(ADC)都含有微管蛋白聚合抑制剂,例如T-DM1和SGN-35(Brentuximab vedotin;参考文献13)。我们合成了一种新型ADC,该ADC含有拓扑异构酶I抑制剂,其作用机制与微管蛋白聚合抑制剂不同,并采用了一种新型的自裂解连接子系统,该系统使用氨基亚甲基(AM)基团。虽然应用于SGN-35(Brentuximab vedotin)和其他几种ADC的其他可裂解连接子系统会释放含氨基的有效载荷,但这种AM自裂解连接子系统能够从DS-8201a中释放含有羟基的DXd。此外,这种新型连接子-有效载荷系统能够降低ADC的疏水性,并有助于提高其药物抗体比(DAR)。 T-DM1采用赖氨酸偶联和不可裂解系统,这与DS-8201a的系统截然不同。DS-8201a在体外和体内均显示出强大的HER2特异性疗效,并且通过药物偶联,其曲妥珠单抗的功能效果与T-DM1相当。此外,DS-8201a在大鼠和食蟹猴中的安全性研究表明,DS-8201a具有良好的耐受性。[3] |
| 分子式 |
C52H56FN9O13
|
|---|---|
| 分子量 |
1034.05195617676
|
| 精确质量 |
1,033.40
|
| 元素分析 |
C, 60.40; H, 5.46; F, 1.84; N, 12.19; O, 20.11
|
| CAS号 |
1599440-13-7
|
| 相关CAS号 |
Exatecan mesylate;169869-90-3;Deruxtecan-d6;2760715-89-5;Deruxtecan-d5;Exatecan mesylate dihydrate;197720-53-9; 171335-80-1; 144008-87-7 (HCl)
|
| PubChem CID |
118305111
|
| 外观&性状 |
White to yellow solid powder
|
| 密度 |
1.48±0.1 g/cm3
|
| 沸点 |
1491.1±65.0 °C
|
| LogP |
-0.4
|
| tPSA |
301Ų
|
| 氢键供体(HBD)数目 |
7
|
| 氢键受体(HBA)数目 |
15
|
| 可旋转键数目(RBC) |
22
|
| 重原子数目 |
75
|
| 分子复杂度/Complexity |
2360
|
| 定义原子立体中心数目 |
3
|
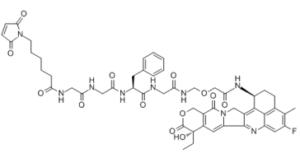
| SMILES |
FC1=CC2=C3C(=C1C)CC[C@@H](C3=C1C(C3=CC4[C@](C(=O)OCC=4C(N3C1)=O)(CC)O)=N2)NC(COCNC(CNC([C@H](CC1C=CC=CC=1)NC(CNC(CNC(CCCCCN1C(C=CC1=O)=O)=O)=O)=O)=O)=O)=O
|
| InChi Key |
WXNSCLIZKHLNSG-MCZRLCSDSA-N
|
| InChi Code |
InChI=1S/C52H56FN9O13/c1-3-52(73)33-19-38-48-31(24-62(38)50(71)32(33)25-75-51(52)72)47-35(14-13-30-28(2)34(53)20-36(60-48)46(30)47)58-43(67)26-74-27-57-41(65)22-56-49(70)37(18-29-10-6-4-7-11-29)59-42(66)23-55-40(64)21-54-39(63)12-8-5-9-17-61-44(68)15-16-45(61)69/h4,6-7,10-11,15-16,19-20,35,37,73H,3,5,8-9,12-14,17-18,21-27H2,1-2H3,(H,54,63)(H,55,64)(H,56,70)(H,57,65)(H,58,67)(H,59,66)/t35-,37-,52-/m0/s1
|
| 化学名 |
Glycinamide, N-[6-(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)-1-oxohexyl]glycylglycyl-L-phenylalanyl-N-[[2-[[(1S,9S)-9-ethyl-5-fluoro-2,3,9,10,13,15-hexahydro-9-hydroxy-4-methyl-10,13-dioxo-1H,12Hbenzo[de]pyrano[3',4':6,7]indolizino[1,2-b]quinolin-1-yl]amino]-2-oxoethoxy]methyl]-
|
| 别名 |
Deruxtecan; DS-8201a; DS8201a; DX-8951 derivative; Trastuzumab deruxtecan; DS 8201a; exatecan derivative; DX 8951; DX8951; Deruxtecan; 1599440-13-7; Mc-ggfg-dxd(1); 5SEB972CO4; Deruxtecan [USAN]; UNII-5SEB972CO4;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years 注意: (1) 该产品在溶液状态不稳定,请现配现用。 (2) 本产品在运输和储存过程中需避光。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~35 mg/mL (~33.85 mM)
H2O : Insoluble (< 1 mg/mL) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 1.75 mg/mL (1.69 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
例如,若需制备1 mL的工作液,可将100 μL 17.5 mg/mL 澄清的 DMSO 储备液加入到400 μL PEG300中,混匀;再向上述溶液中加入50 μL Tween-80,混匀;然后加入450 μL 生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 1.75 mg/mL (1.69 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 17.5mg/mL澄清的DMSO储备液加入到900μL 20%SBE-β-CD生理盐水中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 1.75 mg/mL (1.69 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 10% DMSO+ 40% PEG300+ 5% Tween-80+ 45% saline: 1.75 mg/mL (1.69 mM) 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.9671 mL | 4.8354 mL | 9.6707 mL | |
| 5 mM | 0.1934 mL | 0.9671 mL | 1.9341 mL | |
| 10 mM | 0.0967 mL | 0.4835 mL | 0.9671 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT04644237 | Active, not recruiting | Drug: Trastuzumab deruxtecan |
Non-Small Cell Lung Cancer | Daiichi Sankyo,Inc. | March 19, 2021 | Phase 2 |
| NCT04619004 | Active, not recruiting | Drug: Patritumab Deruxtecan (Fixed dose) |
Non-Small Cell Lung Cancer Metastatic |
Daiichi Sankyo,Inc. | February 2, 2021 | Phase 2 |
| NCT05458401 | Recruiting | Drug: Trastuzumab deruxtecan |
HER2-positive Breast Cancer | Daiichi Sankyo,Inc. | November 11, 2022 |
 DBCO-HS-PEG2-VA-PABC-SG3199
DBCO-HS-PEG2-VA-PABC-SG3199
 Mal-Val-Lys-PAB-MMAE
Mal-Val-Lys-PAB-MMAE
 Maleimide-Ph(3,5-F)-PEG4-Val-Ala-MF-6
Maleimide-Ph(3,5-F)-PEG4-Val-Ala-MF-6
 TL033
TL033
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
