| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
BCL6 (IC50 ~35 μM)
|
|---|---|
| 体外研究 (In Vitro) |
将 DLBCL 细胞暴露于 50 μM FX1 30 分钟。所有三个 BCL6 靶基因在 FX1 的 BCOR 和 SMRT 募集中均显着减少,但在阴性对照基因座上则没有。 BCL6 阴性 DLBCL 细胞系不受 FX1 影响,在这些位点几乎没有 SMRT。当 FX1 和 79-6 在用 50 μM FX1 处理 6 小时后的 DLBCL 细胞中进行定量 ChIP 测定时,很明显,FX1 在阻止 BCL6 与 SMRT 结合方面比 79-6 具有更大的效力。 FX1 暴露于 DLBCL 细胞后的四个连续时间点收集 mRNA。在两个独立的 DLBCL 细胞系中,与媒介物相比,FX1 几乎总是显着诱导这些基因的去抑制[1]。
|
| 体内研究 (In Vivo) |
FX1 治疗小鼠的脾脏在宏观上与载体对照小鼠没有区别。根据流式细胞术测定,TFX1 对 B 细胞的总体丰度没有影响。当暴露于 FX1 时,GC B 细胞 (GL7+FAS+B220+) 显着减少。 IHC 分析脾脏的结构。用 B220 抗体染色后可以看到正常的 B 细胞滤泡结构,但用 GC 特异性 B 细胞标记物花生凝集素染色后可以看到 GC 的显着损失。半衰期被认为是 12 小时左右。最后,确定FX1是否会对小鼠产生毒性作用。与媒介物相比,用 FX1 处理的小鼠的肺、消化道、心脏、肾脏、肝脏、脾脏和骨髓的 H&E 染色切片没有显示出毒性、炎症或感染的迹象[1]。
FX1表型在体内复制BCL6 BTB结构域表型。[1] BCL6 BTB协同抑制因子结合位点的突变导致正常的B细胞发育,但在小鼠中GC形成的严重丧失,在T细胞依赖免疫后仅形成少量残留的GC(例如,如图3A和ref. 22所示)。为了确定FX1是否能够再现这种表型,研究人员用羊红细胞(一种T细胞依赖性抗原)免疫C57BL/6小鼠,然后用每日剂量的80 mg/kg或载体FX1治疗它们。治疗10天后(GCs正常达到峰值),对小鼠实施安乐死并取脾。fx1处理小鼠的脾脏在宏观上与对照没有区别(图3A)。正如预期的那样,流式细胞术测量的总B细胞丰度不受FX1的影响(图3B)。相反,与BCL6 BTB突变表型相似,暴露于FX1后,GC B细胞(GL7+FAS+B220+)显著减少(P < 0.0001;图3 c)。研究人员还通过免疫组化检查了脾结构。B220抗体染色显示正常的B细胞滤泡结构,而GC B细胞特异性标记物花生凝集素染色显示GC严重缺失(图3D)。这种缺陷进一步表现为GCs数量的显著减少(P = 0.0001)和GCs占据的脾表面积(2.4倍;P < 0.001),与车辆对照组相比(图3D)。通过Ki-67染色也可以明显看到GC B细胞增殖的丧失,这表明FX1-处理小鼠中Ki-67+ GC结构的丧失(图3D)。 FX1在体外和体内均能有效抑制bcl6依赖性GCB-DLBCLs。[1] FX1的目的是杀死bcl6依赖性肿瘤。BCL6在dlbcl的GCB类中高表达。根据是否受BCL6敲低或抑制的影响,先前已将DLBCL细胞系分为BCL6依赖型或独立型(34)。为了评估FX1抑制dlbcl的能力,研究人员用不同浓度的FX1处理了一组GCB-DLBCL细胞系(8个依赖于BCL6, 4个不依赖于BCL6) 48小时,并确定了与载体处理细胞(GI50)相比,抑制50%生长所需的化合物浓度。FX1对bcl6依赖性dlbcl表现出选择性作用,平均GI50值约为36 μM,而在bcl6非依赖性dlbcl中无法确定GI50值,因为在高于125 μM的药物浓度下,它们的生长抑制甚至达不到50%(图4A)。值得注意的是,一些被定义为BCL6不依赖型的DLBCL细胞系仍然对BCL6抑制剂保持一定程度的反应性(34)。BCL6依赖性丧失发生的机制尚不清楚,尽管在某些情况下(例如,托莱多细胞),这伴随着BCL6表达几乎完全丧失(补充图4A)。[1] 研究人员接下来希望在体内评估FX1的抗淋巴瘤活性。然而,研究人员首先评估了该化合物的基本药代动力学。16只移植SUDHL-6的SCID小鼠经腹腔注射FX1 50 mg/kg,连续采集血清和组织标本。观察FX1维持50 μM的血清浓度10小时(补充图4B)。半衰期估计约为12小时(补充图4B)。从这些小鼠的肿瘤组织中提取mRNA,评估BCL6靶基因CD69、CDKN1A和CXCR4的丰度。靶基因的最大诱导发生在4-6小时,随后下降(补充图4C)。最后,研究人员评估了FX1是否会对小鼠产生毒性作用。BCL6 BTB突变小鼠没有器官功能障碍或疾病,因此研究人员不会期望靶毒性(22)。一组10只BALB/c小鼠,每天给药100 mg/kg/d FX1治疗10天,之后处死动物,分析器官损伤的组织学证据(补充图4D和补充表2)。与对照组相比,FX1治疗小鼠的肺、胃肠道、心脏、肾脏、肝脏、脾脏和固定器官骨髓的h&e染色切片未见明显的毒性、炎症或感染迹象。研究人员还检测了fx1处理小鼠的外周血计数和血清化学,所有这些参数都保持在正常范围内(补充表2)。 为了确定FX1与79-6在体内的疗效,研究人员首先在SCID小鼠中使用OCI-Ly7 DLBCL细胞建立了DLBCL异种移植物。当肿瘤体积达到约100 mm3时,开始使用每日25 mg/kg或50 mg/kg的FX1或79-6或载药。当对照物达到最大允许肿瘤负荷时(第10天)处死动物。引人注目的是,FX1引起dlbcl的深刻而显著的抑制(P = 0.001;图4B),确实不仅阻止了异种移植物的生长,而且还使这些肿瘤从其初始体积缩小。较低的25 mg/kg剂量已达到最大效果。相比之下,79-6在这些剂量下表现出更弱的抗肿瘤活性(25 mg/kg时肿瘤生长减少42%,50 mg/kg时肿瘤生长减少64%)(图4C)。TUNEL和Ki-67染色显示,FX1也比79-6诱导更多的细胞凋亡和生长停滞(补充图4E)。在使用SUDHL-6 DLBCL细胞的第二种异种移植模型中,FX1的这种强效剂量依赖性抗淋巴瘤作用得到了证实(补充图4、F和G)。为了证实抗肿瘤作用是淋巴瘤细胞自主的,而不是由于对宿主的影响,研究人员还测试了FX1对不依赖bcl6的托莱多异种移植物的作用。暴露于50mg /kg FX1对这些异种移植物没有影响(补充图4,H-J)。[1] 为了验证ABC-DLBCL在体内也可以被FX1靶向,研究人员使用悬浮在Matrigel s.c.中的HBL-1 ABC-DLBCL细胞系在NOD/SCID小鼠中产生异种移植物。当肿瘤达到约100 mm3时,研究人员用每天50 mg/kg的FX1剂量治疗小鼠。治疗10天后,研究人员观察到,暴露于50 mg/kg FX1的小鼠的肿瘤体积减少了约70%(图5、B和C)。研究人员还观察到,与用载药治疗的肿瘤相比,FX1治疗的肿瘤中TUNEL染色的凋亡细胞从2.5%增加到10%(图5D)。[1] |
| 酶活实验 |
MST测量。[1]
重组BCL6 BTB使用RED-NHS标记试剂盒进行标记。根据制造商的说明,在随附的标记缓冲液中使用浓度为20 μM的蛋白质(摩尔染料/蛋白质比为2:1),在室温下进行标记反应30分钟。用用PBS缓冲液(PBS, 0.005%吐温-80)平衡的染料去除柱去除未反应的染料。标记/蛋白比采用650 nm光度法和Bradford试剂测定。因此,比率通常达到0.8。标记的BCL6 BTB用添加0.05%吐温-80的PBS缓冲液调整至400 nM。将SMRT、79-6和FX1溶解在添加0.05% Tween-80和10% DMSO的PBS缓冲液中,在相同的缓冲液中制备16个1:1的稀释液,得到的配体浓度从19 pM到625 μM不等。对于热泳,每种配体稀释剂与1体积标记的BCL6 BTB混合,使荧光标记的BCL6 BTB最终浓度为200 nM,最终配体浓度为9 pM至312 nM,最终浓度为5% DMSO最终浓度。孵育10分钟后,将每种溶液约4 μl填充到Monolith NT标准处理毛细血管中。使用Monolith NT.115仪器在温度为25°C的条件下,分别以5秒/30秒/5秒激光开/关时间测量热泳动。以90%的发光二极管(LED)功率和40%的MST功率调节仪器参数。利用热电泳信号对3个独立实验的数据进行分析。 核磁共振实验。[1] 所有核磁共振实验均在30°C下使用配备低温探针的布鲁克600 MHz光谱仪进行记录。BCL6 BTB结构域的分配基于200 μM 13C、15N BCL6的HNCA和HNCOCA实验记录以及先前发表的分配。BCL6 BTB侧槽处的Fo-Fc电子密度图绘制为2.0 σ水平。由于占用率不足,综合体中1085的整体结构改进无法可靠地完成。 |
| 细胞实验 |
使用 BCL6、SMRT、BCOR 或 IgG 对照抗体,在 DLBCL 细胞中暴露于 FX1(黑条)或媒介物(白条)的 SUDHL-6 细胞中进行定量 ChIP,以富集细胞中已知的 BCL6 结合位点。 CD69、CXCR4 和 DUSP5 基因座,以及阴性对照区。
FX1联合阿霉素。[1] 对于联合治疗,我们将细胞暴露于每种药物单独或以恒定比例组合的剂量曲线中,并用CellTiter-Blue测定细胞活力。为了比较不同的处理方案,我们将细胞分为三个重复处理:FX1和阿霉素同时处理,细胞处理48小时;FX1,第一次和24小时后加阿霉素治疗48小时;先用阿霉素治疗,24小时后加FX1治疗48小时。然后利用CompuSyn软件绘制剂量效应曲线,计算剂量减少指数。 原代细胞处理。[1] 将淋巴结活检的单细胞悬液解冻后,在添加20%人血清、谷氨酰胺(2X)、甘氨酸(5mm)、青霉素和链霉素的Advanced RPMI中重悬。台盼蓝计数法测定细胞数量和活力。在添加10%牛血清和青霉素、链霉素的DMEM中培养的HK细胞(2000 rad)在37℃下粘附在组织培养板上。抽吸培养基,将患者样品镀在HK喂料层上。将样品暴露于FX1或载体中48小时,并通过用PerCP cy5.5偶联抗cd3和fitc偶联抗cd20以及膜联蛋白V和DAPI染色分析细胞活力。膜联蛋白V/DAPI双阴性的CD20+CD3 -细胞被认为是活的恶性B细胞。主要样本按Hans IHC标准分类。 |
| 动物实验 |
携带SUDHL-6异种移植瘤的SCID小鼠
50 mg/kg 腹腔注射 将6至8周龄的雄性SCID小鼠皮下注射10~7代低代人SUDHL-6、OCI-Ly7或Toledo细胞。或者,将6至8周龄的NOD/SCID小鼠注射低代HBL-1细胞。当肿瘤达到可触及的大小(约100 mm³)时,将小鼠随机分配到治疗组,并腹腔注射25或50 mg/kg/d的药物。药物用PEG-400溶解,并储存于-20°C直至使用。每周使用电子游标卡尺测量肿瘤大小3次,并根据以下公式计算肿瘤体积:肿瘤体积(mm³)=(最小直径² × 最大直径)/2。数据以均值±标准误表示,采用Mann-Whitney检验,P<0.05时认为差异具有统计学意义。[1] |
| 参考文献 | |
| 其他信息 |
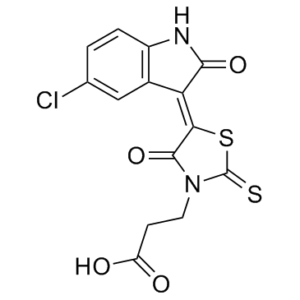
FX1 属于吲哚酮类化合物,其结构为 5-氯吲哚酮,其中 3 位亚甲基氢被 N-(2-羧乙基)罗丹宁-5-亚基取代。它具有抗肿瘤活性。FX1 属于吲哚酮类化合物、有机氯化合物、噻唑烷酮类化合物和单羧酸类化合物。其功能与 3-亚甲基吲哚酮和罗丹宁类似。
弥漫性大 B 细胞淋巴瘤 (DLBCL) 起源于增殖的 B 细胞经历生发中心反应的不同阶段。在活化 B 细胞 DLBCL (ABC-DLBCL) 中,这是一类对现有疗法反应不佳的 DLBCL,染色体易位和扩增导致 B 细胞淋巴瘤 6 (BCL6) 癌基因的组成型表达。 BCL6在维持这些淋巴瘤中的作用尚未得到研究。在此,我们设计了对BCL6具有比其内源性辅阻遏物配体更高亲和力的小分子抑制剂,以评估其靶向ABC-DLBCL的治疗效果。我们采用一种名为SILCS的计算机辅助药物设计功能基团映射方法,构建了一种特异性BCL6抑制剂FX1。FX1的效力比内源性辅阻遏物高10倍,并能结合BCL6侧沟的关键区域。FX1破坏了BCL6抑制复合物的形成,重新激活了BCL6靶基因,并模拟了表达带有辅阻遏物结合位点突变的BCL6基因工程小鼠的表型。低剂量FX1可诱导携带DLBCL异种移植瘤的小鼠体内已建立的肿瘤消退。此外,FX1在体外和体内均能抑制ABC-DLBCL细胞,并在离体实验中抑制了原代人ABC-DLBCL标本的增殖。这些研究结果表明,ABC-DLBCL 是一种 BCL6 依赖性疾病,可通过合理设计的、亲和力高于天然 BCL6 配体的抑制剂进行靶向治疗。[1] 在许多细胞系中,对 BCL6 抑制剂的反应似乎与 BCL6 水平相关,即表达 BCL6 水平较高的细胞系需要更高剂量的抑制剂才能抑制。例如,OCI-Ly1 细胞表达高水平的 BCL6,因此需要比表达 BCL6 水平较低的 SUDHL-6 和 DOHH-2 细胞更多的化合物。然而,这条规则存在许多例外,这可能是由于不同的体细胞突变组合和其他可能改变 BCL6 功能的因素所致。对原发性 DLBCL 的研究受到限制,因为这些肿瘤无法在体外培养足够长的时间以充分评估 FX1 的治疗效果。然而,我们对原代标本的研究证实了细胞系数据,表明GCB型和ABC型弥漫性大B细胞淋巴瘤(DLBCL)细胞依赖于BCL6,并且对BCL6抑制剂敏感。此外,FX1可以增强GCB型和ABC型DLBCL对细胞毒性疗法的反应。因此,BCL6靶向疗法与标准抗淋巴瘤药物联合应用可能产生更优的临床疗效,这对于更容易出现化疗耐药性的高危患者尤为重要。因此,我们的研究将可能对BCL6靶向疗法有反应的淋巴瘤范围扩大到包括GCB型和ABC型DLBCL。鉴于ABC型DLBCL的预后较差,这一点尤为重要。我们期待利用SILCS进一步优化FX1并生成其他化学骨架,从而获得能够使这些难治性患者获益的化合物。[1] |
| 分子式 |
C14H9CLN2O4S2
|
|
|---|---|---|
| 分子量 |
368.8153
|
|
| 精确质量 |
367.969
|
|
| 元素分析 |
C, 45.59; H, 2.46; Cl, 9.61; N, 7.60; O, 17.35; S, 17.39
|
|
| CAS号 |
1426138-42-2
|
|
| 相关CAS号 |
|
|
| PubChem CID |
135886621
|
|
| 外观&性状 |
Red solid powder
|
|
| LogP |
1.4
|
|
| tPSA |
148
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
23
|
|
| 分子复杂度/Complexity |
808
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
C1=CC2=NC(=O)C(=C2C=C1Cl)C3=C(N(C(=S)S3)CCC(=O)O)O
|
|
| InChi Key |
JYBGCTWNOMSQJY-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C14H9ClN2O4S2/c15-6-1-2-8-7(5-6)10(12(20)16-8)11-13(21)17(14(22)23-11)4-3-9(18)19/h1-2,5,21H,3-4H2,(H,18,19)
|
|
| 化学名 |
3-[5-(5-chloro-2-oxoindol-3-yl)-4-hydroxy-2-sulfanylidene-1,3-thiazol-3-yl]propanoic acid
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1 mg/mL (2.71 mM) (饱和度未知) in 10% DMSO + 40% PEG300 +5% Tween-80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 10.0 mg/mL澄清的DMSO储备液加入到400 μL PEG300中,混匀;再将50 μL Tween-80+加入到上述溶液中,混匀;然后加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7113 mL | 13.5567 mL | 27.1135 mL | |
| 5 mM | 0.5423 mL | 2.7113 mL | 5.4227 mL | |
| 10 mM | 0.2711 mL | 1.3557 mL | 2.7113 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|---|
|
|
 RMC-5127
RMC-5127
 Degarelix acetate hydrate
Degarelix acetate hydrate
 DDO-2728
DDO-2728
 Vatalanib hydrochloride (Vatalanib hydrochloride; PTK787 hydrochloride; ZK-222584 hydrochloride; CGP-797870 hydrochloride)
Vatalanib hydrochloride (Vatalanib hydrochloride; PTK787 hydrochloride; ZK-222584 hydrochloride; CGP-797870 hydrochloride)
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA



 463611831
463611831
