| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
GnRHR/LHRHR; gonadotropin-releasing hormone receptor (IC50 = 3 nM)
|
|---|---|
| 体外研究 (In Vitro) |
与其他 LHRH 拮抗剂(如 Ganirelix (HY-P1628)、Abarelix (HY-13534) 和 Cetrorelix (HY-P0009)[1])相比,地加瑞克释放组胺的能力最低,且组胺释放特性非常弱。
除 PC-3 细胞外,地加瑞克 (1 nM-10 μM,0-72 小时) 可降低所有前列腺细胞系的细胞活力,包括 WPE1-NA22、WPMY-1、BPH-1 和 VCaP 细胞 [2]。 通过细胞凋亡,地加瑞克 (10 μM,0-72 小时) 直接影响前列腺细胞的生长 [2]。 |
| 体内研究 (In Vivo) |
在阉割大鼠中,地加瑞克(0-10 μg/kg;皮下注射;一次)以剂量依赖性方式降低血浆睾酮和 LH 水平 [3]。
当在冷冻保存的肝细胞和源自动物肝组织的微粒体中孵育时,地加瑞克保持稳定。在狗和大鼠中,大部分地加瑞克剂量在 48 小时内通过尿液和粪便以等量排出(每种基质中为 40-50%);然而,在猴子中,主要排泄途径是肾脏(22%)和粪便(50%)[4]。 |
| 细胞实验 |
细胞活力测定 [2]
细胞系:VCaP、LNCaP、BPH-1、WPMY-1 和 WPE1-NA22 百分比:1 nM-10 μM 孵育时间:WPMY-1 细胞 48 和 72 小时,WPE1-NA22 细胞 72 小时,BPH-1 细胞 48 和 72 小时,LNCaP 细胞 48 和 72 小时 结果:除 PC-3 细胞外,所有前列腺细胞系的细胞活力均降低。 细胞凋亡分析[2] 细胞系:WPE1-NA22、BPH-1、LNCaP 和 VCaP 浓度:10 μM 孵育时间:24、48 和 72 小时 结果:诱导 caspase 3/7 活化显著增加。 |
| 动物实验 |
动物模型:去势雄性Sprague-Dawley大鼠[3]
剂量:0.3、1、3和10 μg/kg或12.5、50和200 μg/kg 给药途径:皮下注射,单次 结果:可使血浆LH水平降低,且该降低呈剂量依赖性,最小有效剂量为3 μg/kg。50 μg/kg和200 μg/kg剂量组的Tmax值分别为1小时和5小时,表观血浆清除半衰期(t1/2)值分别为12小时和67小时,吸收半衰期(t1/2)值分别为4分钟和30分钟。 最小有效剂量为1 μg/kg,并可引起血浆睾酮水平呈剂量依赖性下降。 |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
皮下注射后,地加瑞克在注射部位形成药物库,药物从该库缓慢释放入血液循环。单次静脉推注2mg/kg后,地加瑞克血浆峰浓度在6小时内达到,浓度为330 ng/mL。Ki = 0.082 ng/mL,93%的受体被完全抑制;平均滞留时间(MRT)= 4.5天。 粪便(70%至80%)和肾脏(20%至30%的药物为原形)排泄。 中央室:8.88 - 11.4 L;外周隔室:40.9 L 前列腺癌患者皮下注射地加瑞克后,清除率约为 9 L/hr。 采用 (3)H-地加瑞克和超速离心技术测定了小鼠、大鼠、犬、猴和人血浆中的蛋白结合率。动物和人体血浆蛋白结合率约为 90%。研究了大鼠、犬和猴分别以 0.03 mg/kg、0.003 mg/kg 和 0.0082 mg/kg 的剂量给予 (3)H-地加瑞克后放射性分布情况。处死动物并进行尸检后,测定了组织放射性。高浓度主要见于皮下注射部位和排泄器官。在内分泌和生殖系统的某些器官中,通常观察到药物浓度较低,但仍高于血浆浓度,这些器官大多含有促性腺激素释放激素(LHRH)的特异性受体;在消除期,在富含网状内皮细胞的器官中也观察到药物浓度较高。未发现组织滞留迹象。 在大鼠、犬和猴中研究了皮下注射(3)H-地加瑞克后放射性平衡的情况。地加瑞克主要以原形经尿液排出,并在动物和人体内经肝胆途径消除的过程中经历肽类降解。 皮下给药后,地加瑞克在注射部位形成局部药物库,导致活性药物的缓释和延长释放。药物从药物库的释放取决于制剂中的浓度和给药体积。此外,在重复给药研究中,增加给药制剂中的药物浓度会导致血浆峰浓度 (Cmax) 和给药间隔内血浆浓度-时间曲线下面积 (AUC) 出现亚比例性增加,血浆谷浓度 (Ctrough) 增加,末端半衰期 (t1/2) 延长,从而延长达到稳态所需的时间,并且达到血浆峰浓度的时间 (Tmax) 有延长的趋势。皮下注射后,地加瑞克在注射部位形成药物储存库,药物从该储存库缓慢释放到血液循环中。皮下注射单次 240 mg 剂量(浓度为 40 mg/mL)后,地加瑞克的血浆峰浓度通常在 2 天内达到。地加瑞克的药代动力学行为受其在注射液中的浓度影响很大。约 90% 的药物与血浆蛋白结合。皮下给药后,未在血浆中检测到具有定量意义的代谢物。体外研究表明,地加瑞克并非细胞色素P-450 (CYP)酶或P-糖蛋白转运系统的底物、诱导剂或抑制剂。地加瑞克的消除呈双相性,在前列腺癌患者中,皮下注射240 mg剂量(浓度为40 mg/mL)后,中位终末半衰期约为53天。地加瑞克在通过肝胆系统时会发生肽水解,主要以肽片段的形式经粪便排出。地加瑞克给药后,约20-30%经肾脏排泄,提示约70-80%经肝胆系统排泄。 有关地加瑞克(共6项)的更多吸收、分布和排泄(完整)数据,请访问HSDB记录页面。 代谢/代谢物 地加瑞克在通过肝胆系统时,70%-80%发生肽水解,然后经粪便排泄。无活性或非活性代谢物,也无CYP450同工酶参与。 在雄性大鼠、豚鼠、兔、狗、猴和人的肝微粒体中,研究了地加瑞克的稳定性,研究时间长达60分钟。在兔、狗、猴和人的肝微粒体中均未检测到地加瑞克的降解。在豚鼠和大鼠的肝微粒体中观察到地加瑞克轻微降解的趋势。进一步在人肝微粒体中研究了地加瑞克长达 60 分钟的体外代谢。据报道,地加瑞克在人和动物中的代谢模式相似。地加瑞克几乎不是氧化代谢的底物,但会被肽酶降解,生成各种截短肽。在人血浆中仅观察到一种代谢物的低浓度,这种代谢物也在大鼠、狗和猴子中检测到。 生物半衰期 末端半衰期:41.5 - 70.2 天;吸收半衰期:32.9 小时;注射部位半衰期:1.17 天。 地加瑞克以双相方式消除,在前列腺癌患者中皮下注射 240 mg 剂量(浓度为 40 mg/mL)后,中位末端半衰期约为 53 天。 |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
地加瑞克治疗与高达三分之一患者的血清酶升高相关。然而,这些升高通常较轻且具有自限性,即使不调整剂量也能自行恢复。ALT 值超过正常值上限 3 倍的患者不到 1%。少数患者因血清酶升高而需要停药,但在地加瑞克的早期临床试验中,未报告出现黄疸或临床表现明显的急性肝损伤的病例。自获批并广泛应用以来,尽管地加瑞克的普遍使用受到限制,但尚未有已发表的临床上明显的肝损伤病例报告。 可能性评分:E(不太可能是临床上明显的肝损伤的原因)。 蛋白结合 90%的药物与血浆蛋白结合。 药物相互作用 由于雄激素剥夺治疗可能延长QTc间期,因此应仔细评估地加瑞克与已知可延长QTc间期的药物或可诱发尖端扭转型室性心动过速的药物(例如IA类(如奎尼丁、丙吡胺)或III类(如胺碘酮、索他洛尔、多非利特、伊布利特)抗心律失常药物、美沙酮、西沙必利、莫西沙星、抗精神病药物等)合用时的安全性。 |
| 参考文献 |
|
| 其他信息 |
治疗用途
地加瑞克用于治疗晚期前列腺癌。/用途详见美国产品标签/ 药物警告 地加瑞克禁用于妊娠期或可能妊娠的女性。孕妇使用地加瑞克可能对胎儿造成伤害。 FDA妊娠风险等级:X /妊娠期禁用。动物或人体研究,或研究性或上市后报告均已证实,胎儿异常或风险明显大于对患者的任何潜在获益。/ 注射部位最常见的不良反应为疼痛(28%)、红斑(17%)、肿胀(6%)、硬结(4%)和结节(3%)。这些不良反应大多为短暂性,程度为轻度至中度,主要发生在起始剂量时,且导致停药的比例很低(<1%)。接受地加瑞克治疗的患者中,3级注射部位反应的发生率≤2%。 共有1325例前列腺癌患者接受了Firmagon治疗,给药方式为每月一次(60-160 mg)或单次给药(最高320 mg)。其中1032例患者(78%)接受了至少6个月的治疗,853例患者(64%)接受了一年或更长时间的治疗。Firmagon治疗期间最常见的不良反应包括注射部位反应(如疼痛、红斑、肿胀或硬结)、潮热、体重增加、疲劳以及血清转氨酶和γ-谷氨酰转移酶(GGT)水平升高。大多数不良反应为 1 级或 2 级,3/4 级不良反应发生率低于 1%。 有关 Degarelix 的更多药物警告(完整)数据(共 15 条),请访问 HSDB 记录页面。 药效学 Degarelix 是 GnRH 十肽的合成衍生物,是 GnRH 受体的配体。内源性 GnRH 与 GnRH 受体结合后,促性腺激素和雄激素的产生。Degarelix 拮抗 GnRH 受体,从而阻断垂体释放 LH 和 FSH。LH 和 FSH 的水平呈浓度依赖性下降。LH 的减少导致睾丸释放的睾酮减少。 |
| 分子式 |
C84H109CLN18O19
|
|---|---|
| 分子量 |
1710.3
|
| 精确质量 |
1708.78049
|
| CAS号 |
934246-14-7
|
| 相关CAS号 |
214766-78-6;Degarelix-d7;934016-19-0;934246-14-7
|
| PubChem CID |
25070695
|
| 序列 |
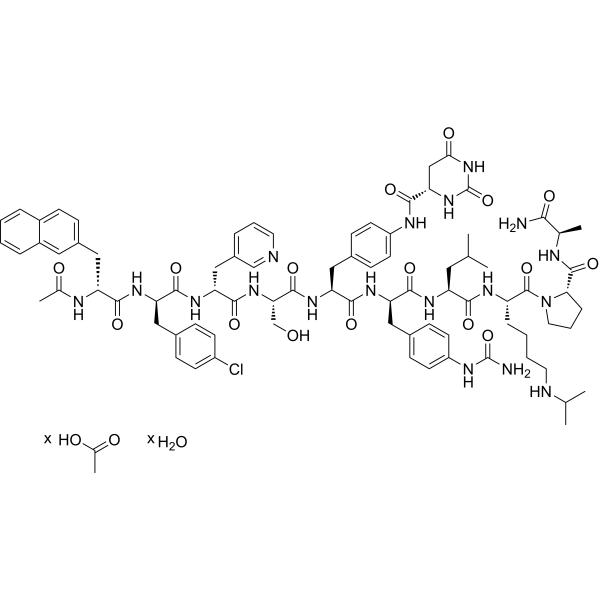
Ac-D-2Nal-D-Phe(4-Cl)-D-3Pal-Ser-Phe(4-S-dihydroorotamido)-D-Phe(4-ureido)-Leu-Lys(iPr)-Pro-D-Ala-NH2.CH3CO2H.H2O
|
| 短序列 |
XXXSXXLXPA
|
| 外观&性状 |
Solid powder
|
| LogP |
0
|
| tPSA |
551 Ų
|
| 氢键供体(HBD)数目 |
19
|
| 氢键受体(HBA)数目 |
21
|
| 可旋转键数目(RBC) |
41
|
| 重原子数目 |
122
|
| 分子复杂度/Complexity |
3420
|
| 定义原子立体中心数目 |
11
|
| SMILES |
ClC1C=CC(=CC=1)C[C@H](C(N[C@H](CC1C=NC=CC=1)C(N[C@@H](CO)C(N[C@@H](CC1C=CC(=CC=1)NC([C@@H]1CC(NC(N1)=O)=O)=O)C(N[C@H](CC1C=CC(=CC=1)NC(N)=O)C(N[C@@H](CC(C)C)C(N[C@@H](CCCCNC(C)C)C(N1CCC[C@H]1C(N[C@@H](C(N)=O)C)=O)=O)=O)=O)=O)=O)=O)=O)NC([C@@H](CC1C=CC2C=CC=CC=2C=1)NC(C)=O)=O.OC(C)=O
|
| InChi Key |
QMBXFMRFTMPFEY-YECCWIQASA-N
|
| InChi Code |
InChI=1S/C82H103ClN18O16.C2H4O2.H2O/c1-45(2)35-60(72(107)92-59(16-9-10-33-87-46(3)4)80(115)101-34-12-17-68(101)79(114)88-47(5)70(84)105)93-74(109)63(38-51-23-30-58(31-24-51)91-81(85)116)95-76(111)64(39-50-21-28-57(29-22-50)90-71(106)66-42-69(104)100-82(117)99-66)97-78(113)67(44-102)98-77(112)65(41-53-13-11-32-86-43-53)96-75(110)62(37-49-19-26-56(83)27-20-49)94-73(108)61(89-48(6)103)40-52-18-25-54-14-7-8-15-55(54)36-52;1-2(3)4;/h7-8,11,13-15,18-32,36,43,45-47,59-68,87,102H,9-10,12,16-17,33-35,37-42,44H2,1-6H3,(H2,84,105)(H,88,114)(H,89,103)(H,90,106)(H,92,107)(H,93,109)(H,94,108)(H,95,111)(H,96,110)(H,97,113)(H,98,112)(H3,85,91,116)(H2,99,100,104,117);1H3,(H,3,4);1H2/t47-,59+,60+,61-,62-,63-,64+,65-,66+,67+,68+;;/m1../s1
|
| 化学名 |
(4S)-N-[4-[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-acetamido-3-naphthalen-2-ylpropanoyl]amino]-3-(4-chlorophenyl)propanoyl]amino]-3-pyridin-3-ylpropanoyl]amino]-3-hydroxypropanoyl]amino]-3-[[(2R)-1-[[(2S)-1-[[(2S)-1-[(2S)-2-[[(2R)-1-amino-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxo-6-(propan-2-ylamino)hexan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-[4-(carbamoylamino)phenyl]-1-oxopropan-2-yl]amino]-3-oxopropyl]phenyl]-2,6-dioxo-1,3-diazinane-4-carboxamide;acetic acid;hydrate
|
| 别名 |
Degarelix acetate hydrate; Firmagon; Degarelix acetate (USAN); Degarelix acetate [USAN]; FE200486;
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
Soluble in DMSO and H2O
|
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.5847 mL | 2.9235 mL | 5.8469 mL | |
| 5 mM | 0.1169 mL | 0.5847 mL | 1.1694 mL | |
| 10 mM | 0.0585 mL | 0.2923 mL | 0.5847 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Stereotactic Body Radiation Therapy Plus Androgen Receptor Pathway Inhibitor and Androgen Deprivation Therapy for Treatment of Metastatic, Recurrent Hormone-Sensitive Prostate Cancer, DIVINE Trial
CTID: NCT06378866
Phase: Phase 2 Status: Recruiting
Date: 2024-11-26
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
