| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
Aldosterone synthase[1]; Baxdrostat is a selective aldosterone synthase (CYP11B2) inhibitor.
Baxdrostat selectively targets aldosterone synthase (cytochrome P450 11B2, mitochondrial; CYP11B2). It exhibits a high selectivity ratio for CYP11B2 over the highly homologous enzyme 11β-hydroxylase (CYP11B1, responsible for cortisol synthesis), with a selectivity factor (SFCYP11B1 IC50/CYP11B2 IC50) of 100. The binding affinity (Ki) of Baxdrostat for human CYP11B2 is 13 nM. For monkey CYP11B2, the Ki is 4 nM. |
|---|---|
| 体外研究 (In Vitro) |
在使用转染了人 CYP11B2 的 G-402 细胞的细胞酶活性测定中,Baxdrostat 显示出强效抑制活性,EC50 为 14 nM。该化合物竞争性抑制醛固酮合成酶,阻断 11-脱氧皮质酮向醛固酮的转化。体外研究证实,与 11β-羟化酶(CYP11B1)相比,Baxdrostat 对醛固酮合成酶表现出高选择性比率,选择性因子约为 100 倍。Baxdrostat 的主要代谢物 CIN-107-M 对醛固酮合成酶的选择性也高于 11β-羟化酶,其两种对映体对醛固酮合成酶的选择性高出 20 倍以上。
|
| 体内研究 (In Vivo) |
在一项针对难治性高血压患者的 2 期试验(BrigHTN)中,Baxdrostat(2 mg 每日一次)显著降低了血压。治疗 12 周时,平均坐位收缩压(SBP)较基线的绝对降幅为 15.7 mmHg,经安慰剂校正后的降幅为 9.8 mmHg。1 mg 剂量组经安慰剂校正后的 SBP 降幅为 8.7 mmHg。在 3 期试验(BaxHTN)中,这些结果得到了确认,约 40% 的患者达到了健康的血压水平(<130 mmHg),而安慰剂组这一比例不到 20%。在非人灵长类动物研究中,Baxdrostat 可降低醛固酮生成,同时不影响皮质醇水平。该药物还显示出剂量依赖性的血浆醛固酮降低以及血浆钠水平轻度降低和钾水平升高。
在健康志愿者中,baxdrostat 呈剂量依赖性降低血浆醛固酮水平(3 mg/天时最高降低约70%),且不影响皮质醇水平,证实其对CYP11B2的选择性优于CYP11B1(皮质醇合酶)[1] 在耐药性高血压患者中,baxdrostat(1 mg、2 mg或10 mg每日一次)显著降低收缩压(10 mg剂量时较安慰剂最大降幅达11.0 mmHg)[2] |
| 酶活实验 |
使用细胞酶活性测定评估了Baxdrostat对人 CYP11B2 和 CYP11B1 的抑制活性。将含有 CMV 启动子控制下的人 CYP11B1 或 CYP11B2 开放阅读框(ORF)以及新霉素抗性标记物的表达质粒转染至 G-402 细胞中。筛选转染后的细胞,并在含 10% 胎牛血清和 400 μg/ml G418 的 McCoy's 5a 改良培养基中,于 37°C、5% CO2/95% 空气条件下维持培养。测定时,将细胞接种于 96 孔板中,在含 2.5% 活性炭处理的胎牛血清和适当浓度底物(CYP11B2 为 0.3-10 μM 11-脱氧皮质酮,CYP11B1 为 11-脱氧皮质醇)的 DMEM/F12 培养基中孵育 16 小时。孵育后,转移上清液等分试样,使用均相时间分辨荧光(HTRF)测定法分析产物浓度(CYP11B2 为醛固酮;CYP11B1 为皮质醇)。
|
| 细胞实验 |
使用稳定表达人 CYP11B2 的 G-402 细胞评估了Baxdrostat的细胞活性。这些细胞在含 10% 胎牛血清和 400 μg/ml G418 的 McCoy's 5a 改良培养基中,于 37°C、5% CO2/95% 空气条件下维持培养。进行细胞酶活性测定时,将细胞接种于 96 孔板中,在含 2.5% 活性炭处理的胎牛血清和 0.3-10 μM 底物 11-脱氧皮质酮的 DMEM/F12 培养基中孵育 16 小时。孵育后,转移上清液等分试样,使用 HTRF 测定法测定预期产物醛固酮的浓度。
|
| 动物实验 |
初步的药理学表征研究是在食蟹猴中进行的,结果显示 Baxdrostat 可降低醛固酮生成,同时不影响皮质醇水平。
|
| 药代性质 (ADME/PK) |
在健康志愿者中进行的 I 期研究表明,巴司他汀在 0.5–10 mg 剂量范围内具有线性药代动力学特征,中位达峰时间 (Tmax) 为 2–4 小时,平均半衰期约为 15–20 小时 [1]
一项交叉研究表明,巴司他汀与二甲双胍之间无显著的药代动力学相互作用 [3] Baxdrostat 口服给药后吸收迅速,在给药后 4 小时内达到血浆峰浓度(Tmax)。该药物的平均血浆半衰期(t1/2)约为 26 至 31 小时,支持每日一次给药。在治疗剂量范围内,血浆暴露量呈剂量比例性增加。在稳态下(每日一次给药第 10 天),Baxdrostat 的暴露量约为单次给药后的 2 至 2.5 倍。第 1 天约有 7% 的剂量以原形经尿液排出,到第 10 天稳态时增至约 32%。主要代谢物 CIN-107-M 基于 Cmax 约占母体化合物的 8-11%,基于 AUC 约占 10-22,但被认为对药效作用贡献不大。Baxdrostat 具有高口服生物利用度和快速吸收特性。 |
| 毒性/毒理 (Toxicokinetics/TK) |
Baxdrostat在健康志愿者和难治性高血压患者中耐受性良好,未报告严重不良事件。部分患者观察到轻度高钾血症[1][2][br>
在健康志愿者中进行的一项 1 期多剂量递增研究中,Baxdrostat 安全且耐受性良好。未报告死亡或严重不良事件。接受 Baxdrostat 治疗的受试者中所有治疗中出现的不良事件均为轻度。常见的轻度不良事件包括头痛、鼻咽炎和腹泻。Baxdrostat 对血浆皮质醇水平无显著影响,证明了其对醛固酮合成酶的选择性。该药物导致血浆钠水平出现轻度剂量依赖性下降,血浆钾水平出现轻度剂量依赖性升高。
|
| 参考文献 |
[1]. Results from a phase 1, randomized, double-blind, multiple ascending dose study characterizing the pharmacokinetics and demonstrating the safety and selectivity of the aldosterone synthase inhibitor baxdrostat in healthy volunteers. Hypertens Res. 2023 Jan;46(1):108-118.
[2]. The selective aldosterone synthase inhibitor Baxdrostat significantly lowers blood pressure in patients with resistant hypertension. Front Endocrinol (Lausanne). 2022 Dec 9;13:1097968. [3]. Results From a Randomized, Open-Label, Crossover Study Evaluating the Effect of the Aldosterone Synthase Inhibitor Baxdrostat on the Pharmacokinetics of Metformin in Healthy Human Subjects. Am J Cardiovasc Drugs. 2023 May;23(3):277-286. |
| 其他信息 |
Baxdrostat 是一种正在研究用于治疗难治性高血压的药物,其作用机制是通过靶向醛固酮过度生成。它对 CYP11B2 的选择性可避免皮质醇缺乏的风险 [1][2]
Baxdrostat(也称为 CIN-107、RO6836191)是一种新型四氢异喹啉衍生物,由阿斯利康开发(通过 2023 年以约 18 亿美元收购 CinCor Pharma 获得)。该药物旨在治疗与醛固酮水平升高相关的疾病,醛固酮是高血压、心血管疾病和肾功能障碍的关键驱动因素。与传统的盐皮质激素受体阻断剂(如螺内酯)可能因皮质醇失调引起内分泌副作用不同,Baxdrostat 对 CYP11B2 相对于 CYP11B1 的高选择性使其能够在不影响皮质醇的情况下降低醛固酮,可能提供更优的安全性。在 2025 年欧洲心脏病学会(ESC)大会上公布并同时发表于《新英格兰医学杂志》的 3 期 BaxHTN 试验结果显示,在背景抗高血压治疗基础上加用 Baxdrostat(1 mg 或 2 mg 每日一次)可带来具有临床意义的 SBP 降低(与安慰剂相比约 9-10 mmHg),该效果持续长达 32 周,且未发现非预期的安全性问题。该药物也正在被评估用于治疗原发性醛固酮增多症,以及与达格列净联合治疗合并高血压的慢性肾脏病。 |
| 分子式 |
C22H25N3O2
|
|---|---|
| 分子量 |
363.45
|
| 精确质量 |
363.194
|
| 元素分析 |
C, 72.70; H, 6.93; N, 11.56; O, 8.80
|
| CAS号 |
1428652-17-8
|
| 相关CAS号 |
(S)-Baxdrostat;1428652-16-7;(Rac)-Baxdrostat;1428652-15-6
|
| PubChem CID |
71535962
|
| 外观&性状 |
Off-white to light yellow solid powder
|
| LogP |
2.2
|
| tPSA |
62.3
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
27
|
| 分子复杂度/Complexity |
566
|
| 定义原子立体中心数目 |
1
|
| SMILES |
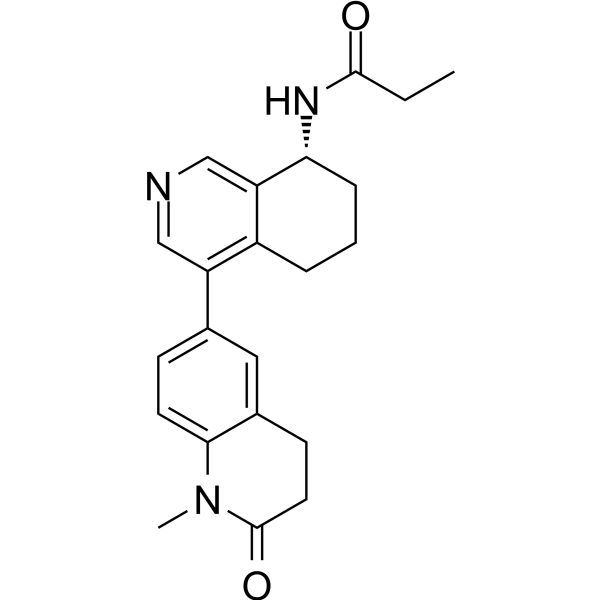
C(N[C@H]1C2=C(CCC1)C(C1C=CC3=C(C=1)CCC(=O)N3C)=CN=C2)(=O)CC
|
| InChi Key |
VDEUDSRUMNAXJG-LJQANCHMSA-N
|
| InChi Code |
InChI=1S/C22H25N3O2/c1-3-21(26)24-19-6-4-5-16-17(12-23-13-18(16)19)14-7-9-20-15(11-14)8-10-22(27)25(20)2/h7,9,11-13,19H,3-6,8,10H2,1-2H3,(H,24,26)/t19-/m1/s1
|
| 化学名 |
N-[(8R)-4-(1-methyl-2-oxo-3,4-dihydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl]propanamide
|
| 别名 |
CIN-107; CIN107; Baxdrostat; Baxdrostat [INN]; NF3P9Z8J5Y; RO6836191; CIN 107
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 100 mg/mL (275.14 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (5.72 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (5.72 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7514 mL | 13.7571 mL | 27.5141 mL | |
| 5 mM | 0.5503 mL | 2.7514 mL | 5.5028 mL | |
| 10 mM | 0.2751 mL | 1.3757 mL | 2.7514 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT06344104
Conditions:Uncontrolled Hypertension|Resistant HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT07007793
Conditions:Primary HyperaldosteronismLink: https://clinicaltrials.gov/ct2/show/NCT06168409
Conditions:Resistant Hypertension
Title:A Study to Evaluate Cortisol Reserve in Response to Adrenocorticotropic Hormone (ACTH) Stimulation Test Following Baxdrostat Treatment Compared to Placebo in Participants With Uncontrolled Hypertension
Status:Completed
updateDate:2025-12-17
Ctid:NCT06336356
Link: https://clinicaltrials.gov/ct2/show/NCT06336356
Conditions:Uncontrolled HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT06034743
Conditions:Uncontrolled Hypertension|Resistant HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT05432167
Conditions:Uncontrolled Hypertension|Chronic Kidney DiseasesLink: https://clinicaltrials.gov/ct2/show/NCT06194032
Conditions:Healthy ParticipantsLink: https://clinicaltrials.gov/ct2/show/NCT06657105
Conditions:Healthy ParticipantsLink: https://clinicaltrials.gov/ct2/show/NCT05459688
Conditions:HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT06357520
Conditions:Healthy ParticipantsLink: https://clinicaltrials.gov/ct2/show/NCT05961384
Conditions:HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT05500820
Conditions:HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT05966324
Conditions:HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT05470725
Conditions:HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT05526690
Conditions:HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT05961397
Conditions:HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT05137002
Conditions:Uncontrolled HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT04519658
Conditions:Resistant HypertensionLink: https://clinicaltrials.gov/ct2/show/NCT01995383
Conditions:AtherosclerosisInvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
