| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
| 靶点 |
VEGFR1 (IC50 = 13 nM); VEGFR2 (IC50 = 4.2 nM); VEGFR3 (IC50 = 46 nM); PDGFRβ (IC50 = 22 nM); Braf (IC50 = 28 nM); VEGFR2 (BRafV600E = 19 nM); Raf-1 (IC50 = 2.5 nM)
|
||
|---|---|---|---|
| 体外研究 (In Vitro) |
Regorafenib 有效抑制 NIH-3T3/VEGFR2 细胞中的 VEGFR2 自磷酸化,IC50 为 3 nM。 Regorafenib 的 IC50 为 90 nM,在 PDGF-BB 刺激后阻断 HAoSMC 中的 PDGFR-β 自磷酸化。罗戈非尼抑制 vegf165 刺激的 HUVEC 增殖,IC50 为 3 nM[1]。瑞戈非尼的 IC50 为 5 μM,以浓度依赖性方式抑制 Hep3B 细胞的生长。 JNK 靶向磷酸化 c-Jun,但不是总 c-Jun,随后在 Hep3B 细胞中被调节剂阿非尼上调[3]。
Regorafenib(0-10 μM,96 小时)在 GIST 882、甲状腺 TT、MDA-MB-231、HepG2、A375 和 SW620 细胞中表现出抗增殖活性[1]。 Regorafenib(BAY 73-4506)是一种新型口服多激酶抑制剂,在生化和细胞激酶磷酸化测定中能有效抑制这些内皮细胞激酶。此外,瑞格非尼还能抑制其他血管生成激酶(VEGFR1/3、血小板衍生生长因子受体β和成纤维细胞生长因子受体1)以及突变致癌激酶KIT、RET和B-RAF。 雷戈非尼以浓度和时间依赖的方式抑制人Hep3B、PLC/PRF/5和HepG2细胞的生长。多种信号通路发生了改变,包括MAP激酶磷酸化ERK和磷酸化JNK及其靶点磷酸化c-Jun。有证据表明FACS导致细胞凋亡、胱天蛋白酶切割和Bax水平升高;以及通过增加Beclin-1和LC3(II)水平来判断自噬的诱导。长时间接触药物会导致细胞静止。与阿霉素化疗不同,药物去除后生长完全恢复。雷戈非尼是一种强效的细胞生长抑制剂。在雷戈非尼治疗中存活的细胞仍然存活,但处于静止状态,在药物去除后能够再生。药物去除后肿瘤细胞生长抑制的可逆性可能具有临床意义。[3] Regorafenib(0–3000 nM,30 分钟)抑制 FGFR 和 pERK1/2 以及 VEGFR2、TIE2 和 PDGFR-β 的自身磷酸化。 Regorafenib 的 IC50 为 5 μM,以浓度依赖性方式抑制 Hep3B 细胞生长。然后,瑞戈非尼会提高 Hep3B 细胞(JNK 靶标)中的磷酸化 c-Jun 水平,但不会提高总 c-Jun 水平[3]。 |
||
| 体内研究 (In Vivo) |
瑞戈非尼在 10 至 100 mg/kg 剂量下可有效减缓 Colo-205 异种移植物的生长,10 mg/kg 剂量下第 14 天的 TGI 为 75%。瑞戈非尼在 MDA-MB-231 模型中,剂量低至 3 mg/kg 时非常有效,产生 81% 的显着 TGI,在剂量为 10 和 30 mg/kg 时,TGI 上升至 93%,达到肿瘤停滞[1]。
Regorafenib(10 mg/kg,口服,每天一次或两次,持续 4 天)可抑制大鼠 GS9L 胶质母细胞瘤模型中的肿瘤生长和肿瘤血管系统[1]。 Regorafenib(0-100 mg/kg,口服,qd × 9)在 Colo-205、MDA-MB-231 和 786-O 模型中表现出抗肿瘤和抗血管生成作用[1]。 动态增强磁共振成像在体内证明了瑞格非尼的抗血管生成作用。雷戈非尼以10mg/kg的剂量口服一次,显著减少了Gadomer在大鼠GS9L胶质母细胞瘤异种移植物血管系统中的外渗。在每日(qd)×4次给药研究中,药效作用在最后一次给药后持续48小时,并与肿瘤生长抑制(TGI)相关。在10和30 mg/kg的qd×5给药后,在人类结直肠异种移植物中观察到肿瘤微血管面积的显著减少。雷戈非尼在小鼠的各种临床前人类异种移植物模型中表现出强烈的剂量依赖性TGI,在乳腺MDA-MB-231和肾786-O癌模型中观察到了肿瘤缩小。乳腺模型的药效学分析显示,增殖标志物Ki-67和磷酸化细胞外调节激酶1/2的染色显著减少。这些数据表明,瑞格非尼是一种耐受性良好、口服活性的多激酶抑制剂,具有独特的靶点特征,可能对人类恶性肿瘤具有治疗益处[1]。 |
||
| 酶活实验 |
体外测试中使用重组 VEGFR2(鼠类 aa785–aa1367)、VEGFR3(鼠类 aa818–aa1363)、PDGFRβ(aa561–aa1106)、Raf-1(aa305–aa648)和 BRafV600E(aa409–aa765)激酶结构域。在恒定的 1 M 瑞戈非尼浓度下,进行初始体外激酶抑制分析。选择响应激酶,例如 VEGFR1 和 RET,用于计算 50% 抑制浓度 (IC50) 值。使用谷胱甘肽-S-转移酶的重组融合蛋白、TIE2 的胞内结构域和肽生物素-Ahx-EPKDDAYPLYSDFG 作为底物,使用均相时间分辨荧光 (HTRF) 测定来测量 TIE2 激酶抑制。
|
||
| 细胞实验 |
GIST 882 和 TT 细胞在含有 L-谷氨酰胺的 RPMI 培养基中生长以进行增殖测定,而 MDA-MB-231、HepG2 和 A375 细胞在始终补充有 10% hiFBS 的 DMEM 中生长。胰蛋白酶处理的细胞以每孔 5×104 个细胞的密度接种在含有含 10% FBS 的完全培养基的 96 孔板中,并在 37°C 下生长过夜。添加载体或瑞格非尼(在完全生长培养基中连续稀释至最终浓度在 10 μM 至 5 nM 之间)和 0.2% DMSO,继续孵育 96 小时。使用 CellTitre-GloTM 测量细胞增殖。 [1]
通过酶联免疫吸附试验(ELISA)和蛋白质印迹分析VEGFR2磷酸化[1] 将转染有人VEGFR2的NIH-3T3细胞以30000个细胞/孔的速度接种在含有10%FBS的Dulbecco改良Eagle培养基的96孔板中;接种后6小时,将培养基换成0.1%BSA/DMEM,继续孵育24小时。细胞在37°C下用0.1%BSA/DMEM/0.1%二甲亚砜(DMSO)中的载体或不同浓度的雷戈非尼处理1小时,然后用终浓度为30 ng/mL的重组VEGF165刺激5分钟。细胞用冷磷酸盐缓冲盐水(PBS)洗涤,并在100μL裂解缓冲液(50 mM HEPES,pH 7.2,1%Triton X-100,1 mM Na3VO4,150 mM NaCl,10%甘油,1.5 mM乙二醇四乙酸和完全蛋白酶抑制剂混合物)中裂解。这是因为。 雷戈非尼治疗[3] 每株细胞系以0.3×105个细胞/2ml含10%FBS的DMEM接种在35mm组织培养皿中。将细胞孵育24小时以允许附着,然后用含有浓度逐渐增加(1μM、2.5μM、5μM、7.5μM和10μM)的雷戈非尼的新鲜培养基替换培养基。在这些实验条件下,允许细胞生长72或96小时。 在Hep3B细胞上进行短期(15、60、180分钟)、中期(24、48、72和96小时)或长期(长达7天)的7.5μMRegorafenib时间过程实验。当细胞被长时间处理时,药物被替换为新鲜的。每个实验都包括一个对照组,其DMSO浓度(溶剂对照组)与用于添加雷戈非尼的浓度相等。每个实验进行三次,重复3次。在特定浓度和孵育时间下进行后续分析。 恢复/可逆性[3] 为了研究停药后细胞增殖的恢复,Hep3B细胞用雷戈非尼5或7.5μM处理3-7天,然后取出培养基,用不含药物的新鲜培养基替换。在不同的后续时间点通过MTT试验评估细胞恢复率。 以0.01、0.05或0.1μM的阿霉素治疗作为阳性对照,研究凋亡过程。 细胞凋亡的FACS分析[3] 按照供应商的规定,使用FITC膜联蛋白V试剂盒检测细胞凋亡。简而言之,收获用不同浓度的雷戈非尼处理48小时的1×106个细胞,并用PBS洗涤。将细胞重新悬浮在结合缓冲液中,然后在5μl AnnexinV-FITC和10μl 7-氨基放线菌素D(7AAD)插入DNA后,在室温下在黑暗中孵育5分钟。将完整细胞与凋亡细胞区分开来。 |
||
| 动物实验 |
|
||
| 药代性质 (ADME/PK) |
吸收、分布和排泄
Cmax = 2.5 μg/mL;Tmax = 4 小时;AUC = 70.4 μgh/mL;稳态 Cmax = 3.9 μg/mL;稳态 AUC = 58.3 μgh/mL;与口服溶液相比,片剂的平均相对生物利用度为 69% 至 83%。 服用 120 mg 放射性标记口服溶液后 12 天内,约 71% 的放射性标记剂量经粪便排出(47% 为母体化合物,24% 为代谢物),19% 的剂量经尿液排出(17% 为葡萄糖醛酸苷)。 瑞戈非尼经肠肝循环,在 24 小时给药间隔内观察到多个血浆浓度峰值。 代谢/代谢物 瑞戈非尼由 CYP3A4 和 UGT1A9 代谢。在人血浆中稳态测定的瑞戈非尼主要循环代谢物为M-2(N-氧化物)和M-5(N-氧化物和N-去甲基),二者均具有与瑞戈非尼相似的体外药理活性和稳态浓度。M-2和M-5的蛋白结合率很高(分别为99.8%和99.95%)。瑞戈非尼是P-糖蛋白抑制剂,而其活性代谢物M-2(N-氧化物)和M-5(N-氧化物和N-去甲基)是P-糖蛋白的底物。 生物半衰期 瑞戈非尼,口服160 mg = 28小时(14-58小时);M-2代谢物,口服160 mg = 25小时(14-32小时);M-5代谢物,口服160 mg = 51小时(32-72小时); |
||
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
在瑞戈非尼的大型临床试验中,血清转氨酶水平升高较为常见,发生率达39%至45%,其中3%至6%的患者转氨酶水平超过正常值上限(ULN)的5倍以上。此外,已有数例瑞戈非尼治疗期间出现临床表现明显的肝损伤的报道,这些损伤通常较为严重,偶有致命性,估计发生率约为0.3%。因此,建议常规监测肝酶。瑞戈非尼引起的肝损伤可表现为多种不同的模式或表型。部分患者在开始服用瑞戈非尼后数日内出现急性肝坏死,血清转氨酶和乳酸脱氢酶水平升高,伴有轻度黄疸,但INR延长并出现肝功能衰竭的体征。这种损伤可能较为严重,但通常具有自限性,且恢复迅速且完全。其他患者表现为类似急性病毒性肝炎的临床模式,伴有肝细胞性(或混合性)血清酶升高和黄疸,黄疸可持续较长时间,且在一些病例中甚至导致死亡。自身免疫和免疫过敏反应并不常见。此外,罕见的瑞戈非尼相关肝损伤病例表现为类似肝窦阻塞综合征或假性肝硬化,伴有明显的肝脏结节和腹水,但最终可改善或消退。最后,瑞戈非尼与其他多激酶抑制剂(舒尼替尼、伊马替尼、索拉非尼)一样,也与高氨血症昏迷发作有关,通常在开始用药后的几天或几周内出现,停药后可迅速逆转。 可能性评分:B(极有可能引起临床上明显的肝损伤)。 妊娠和哺乳期影响 ◉ 哺乳期用药概述 目前尚无瑞戈非尼在哺乳期临床应用的信息。由于瑞戈非尼与血浆蛋白的结合率高达99.5%,因此其在乳汁中的含量可能很低。然而,其一种代谢物的半衰期长达70小时,可能会在婴儿体内蓄积。制造商建议在瑞戈非尼治疗期间以及末次给药后 2 周内停止母乳喂养。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对泌乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白结合 瑞戈非尼与人血浆蛋白的结合率很高 (99.5%)。 |
||
| 参考文献 |
|
||
| 其他信息 |
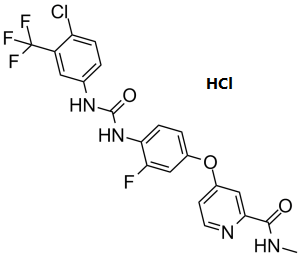
瑞戈非尼是一种吡啶甲酰胺类化合物,由4-[4-({[4-氯-3-(三氟甲基)苯基]氨基甲酰基}氨基)-3-氟苯氧基]吡啶-2-羧酸与甲胺缩合而成。用于治疗既往接受过化疗、抗EGFR或抗VEGF治疗的转移性结直肠癌患者。它具有抗肿瘤、酪氨酸激酶抑制和肝毒性等作用。它是一种芳香醚、吡啶甲酰胺、单氯苯类、(三氟甲基)苯类、单氟苯类和苯脲类化合物。
瑞戈非尼是一种口服的多激酶抑制剂,用于治疗转移性结直肠癌、晚期胃肠道间质瘤和肝细胞癌。瑞戈非尼于2012年9月27日获得FDA批准。2017年4月,瑞戈非尼的适应症扩展至治疗肝细胞癌。 无水瑞戈非尼是一种激酶抑制剂。其作用机制包括作为激酶抑制剂、细胞色素P450 2C9抑制剂、乳腺癌耐药蛋白抑制剂、UGT1A9抑制剂和UGT1A1抑制剂。 瑞戈非尼是一种口服多激酶抑制剂,用于治疗难治性转移性结直肠癌、肝细胞癌和胃肠道间质瘤。瑞戈非尼治疗期间常伴有血清转氨酶升高,并有罕见但有时严重甚至致命的临床表现明显的肝损伤病例。 无水瑞戈非尼是瑞戈非尼的无水形式,瑞戈非尼是一种口服生物利用度高的小分子药物,具有潜在的抗血管生成和抗肿瘤活性。瑞戈非尼可与血管内皮生长因子受体(VEGFR)2和3以及Ret、Kit、PDGFR和Raf激酶结合并抑制其活性,从而抑制肿瘤血管生成和肿瘤细胞增殖。VEGFR是受体酪氨酸激酶,在肿瘤血管生成中发挥重要作用;受体酪氨酸激酶RET、KIT和PDGFR以及丝氨酸/苏氨酸特异性Raf激酶参与肿瘤细胞信号传导。 瑞戈非尼是瑞戈非尼的水合物形式,是一种口服生物利用度高的小分子药物,具有潜在的抗血管生成和抗肿瘤活性。瑞戈非尼可与血管内皮生长因子受体(VEGFR)2和3以及Ret、Kit、PDGFR和Raf激酶结合并抑制其活性,从而抑制肿瘤血管生成和肿瘤细胞增殖。VEGFR是受体酪氨酸激酶,在肿瘤血管生成中发挥重要作用;受体酪氨酸激酶RET、KIT和PDGFR以及丝氨酸/苏氨酸特异性Raf激酶参与肿瘤细胞信号传导。 另见:瑞戈非尼一水合物(活性成分)。 药物适应症 瑞戈非尼适用于治疗既往接受过氟尿嘧啶、奥沙利铂和伊立替康类化疗、抗VEGF治疗以及(如果KRAS野生型)抗EGFR治疗的转移性结直肠癌(CRC)患者。瑞戈非尼也适用于治疗既往接受过甲磺酸伊马替尼和苹果酸舒尼替尼治疗的局部晚期、不可切除或转移性胃肠道间质瘤(GIST)患者。瑞戈非尼也适用于治疗既往接受过索拉非尼治疗的肝细胞癌 (HCC) 患者。 FDA 标签 Stivarga 适用于以下成年患者的单药治疗:既往接受过或不适合接受现有疗法(包括氟尿嘧啶类化疗、抗 VEGF 疗法和抗 EGFR 疗法)治疗的转移性结直肠癌 (CRC) 患者;既往接受过伊马替尼和舒尼替尼治疗后病情进展或不耐受的不可切除或转移性胃肠道间质瘤 (GIST) 患者;既往接受过索拉非尼治疗的肝细胞癌 (HCC) 患者。 治疗所有恶性肿瘤(造血和淋巴组织肿瘤除外) 作用机制 瑞戈非尼是一种小分子抑制剂,可抑制多种膜结合和细胞内激酶,这些激酶参与正常的细胞功能以及肿瘤发生、肿瘤血管生成和肿瘤微环境维持等病理过程。在体外生化或细胞实验中,瑞戈非尼及其主要的人源活性代谢物M-2和M-5在临床应用浓度下可抑制RET、VEGFR1、VEGFR2、VEGFR3、KIT、PDGFR-α、PDGFR-β、FGFR1、FGFR2、TIE2、DDR2、TrkA、Eph2A、RAF-1、BRAF、BRAFV600E、SAPK2、PTK5和Abl的活性。在体内模型中,瑞戈非尼在大鼠肿瘤模型中表现出抗血管生成活性,并在包括一些人类结直肠癌在内的多种小鼠异种移植模型中表现出抑制肿瘤生长和抗转移活性。 |
| 分子式 |
C₂₁H₁₆CL₂F₄N₄O₃
|
|---|---|
| 分子量 |
519.28
|
| 精确质量 |
518.053
|
| CAS号 |
835621-07-3
|
| 相关CAS号 |
Regorafenib;755037-03-7;Regorafenib monohydrate;1019206-88-2
|
| PubChem CID |
11167602
|
| 外观&性状 |
White to off-white solid
|
| LogP |
6.968
|
| tPSA |
95.84
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
33
|
| 分子复杂度/Complexity |
686
|
| 定义原子立体中心数目 |
0
|
| SMILES |
Cl.O=C(NC1C(F)=CC(OC2C=C(C(NC)=O)N=CC=2)=CC=1)NC1C=C(C(F)(F)F)C(Cl)=CC=1
|
| InChi Key |
ACSWJKPZXNIVMY-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C21H15ClF4N4O3.ClH/c1-27-19(31)18-10-13(6-7-28-18)33-12-3-5-17(16(23)9-12)30-20(32)29-11-2-4-15(22)14(8-11)21(24,25)26;/h2-10H,1H3,(H,27,31)(H2,29,30,32);1H
|
| 化学名 |
4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]-3-fluorophenoxy]-N-methylpyridine-2-carboxamide;hydrochloride
|
| 别名 |
BAY-734506 HCl; BAY 734506; BAY734506; Regorafenib HCl. Brand name: Stivarga
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2 mg/mL (3.85 mM) in 50% PEG300 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
*生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 30% PEG400+0.5% Tween80+5% Propylene glycol : 30mg/mL 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9257 mL | 9.6287 mL | 19.2574 mL | |
| 5 mM | 0.3851 mL | 1.9257 mL | 3.8515 mL | |
| 10 mM | 0.1926 mL | 0.9629 mL | 1.9257 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03992456 | Active Recruiting |
Drug: Regorafenib Biological: Panitumumab |
Metastatic Colon Adenocarcinoma Metastatic Colorectal Carcinoma |
Academic and Community Cancer Research United |
April 24, 2020 | Phase 2 |
| NCT04776148 | Active Recruiting |
Drug: regorafenib Drug: lenvatinib |
Colorectal Neoplasms | Merck Sharp & Dohme LLC | March 29, 2021 | Phase 3 |
| NCT03563157 | Active Recruiting |
Drug: Oxaliplatin Drug: Regorafenib |
mCRC Colorectal Cancer Metastatic |
ImmunityBio, Inc. | May 25, 2018 | Phase 1 Phase 2 |
| NCT02788006 | Completed | Drug: Regorafenib 160 mg | Colorectal Adenocarcinoma | Federation Francophone de Cancerologie Digestive |
January 2016 | Phase 2 |
A Study of Continued Treatment With Regorafenib in Participants With Solid Tumors Who Have Participated in Other Bayer Studies
CTID: NCT06246643
PhasePhase 2 Status: Active, not recruiting
Date: 2024-11-20
|
|---|
Regorafenib inhibits key kinase targets in cells expressing VEGFR2, TIE2, PDGFR‐β, or FGFR.Int J Cancer.2011 Jul 1;129(1):245-55. |
Regorafenib inhibits tumor vasculature and tumor growth in a rat GS9L glioblastoma model: time‐course analysis by DCE‐MRI.Int J Cancer.2011 Jul 1;129(1):245-55. |
Regorafenib significantly reduces tumor MVA in the Colo‐205 CRC xenograft model.Int J Cancer.2011 Jul 1;129(1):245-55. |
|---|
Regorafenib exhibits antitumorigenic and antiangiogenic effects in the MDA‐MB‐231 breast xenograft model.Int J Cancer.2011 Jul 1;129(1):245-55. |
In vivoantitumor efficacy of regorafenib.Int J Cancer.2011 Jul 1;129(1):245-55. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
")
")
")
")
")
")
 463611831
463611831
