| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
S-phase kinase-associated protein 2 (Skp2) [1]
S-phase kinase-associated protein 2 (Skp2) [2] S-phase kinase-associated protein 2 (Skp2) [3] S-phase kinase-associated protein 2 (Skp2) (interacts with Skp2/Cks1 E3 ligase complex to block p27 binding,) [4] |
|---|---|
| 体外研究 (In Vitro) |
体外活性:Skp2抑制剂C1处理MCF-7乳腺癌细胞系并没有增加,而是减少了G1期细胞的百分比,同时增加了G2/M细胞群。用 C1(5 μM,16 小时)处理的 T47D 细胞显示 G1 期增加 (p < 0.0001) 和 S 期减少 (p < 0.0001),与 p27 蛋白诱导相关。相反,MCF-7 细胞对 C1 的反应是 G1 期显着减少(35%,p < 0.0001)和 G2-M 期增加(43%,p < 0.0001)。这种 G1 减少和 G2/M 停滞与 C1 剂量相关(图 5C 右侧;分别为 p < 0.001 和 p < 0.01),并且与 p27 蛋白水平增加相关。激酶测定:Skp2抑制剂C1处理MCF-7乳腺癌细胞系并没有增加,而是减少了G1期细胞的百分比,同时增加了G2/M细胞群。用 C1(5 μM,16 小时)处理的 T47D 细胞显示 G1 期增加 (p < 0.0001) 和 S 期减少 (p < 0.0001),与 p27 蛋白诱导相关。相反,MCF-7 细胞对 C1 的反应是 G1 期显着减少(35%,p < 0.0001)和 G2-M 期增加(43%,p < 0.0001)。这种 G1 减少和 G2/M 停滞与 C1 剂量相关(图 5C 右侧;分别为 p < 0.001 和 p < 0.01),并且与 p27 蛋白水平增加相关。细胞测定:用每种抑制剂 (10 μM) 或 0.1% DMSO(媒介物)预处理 501 Mel 细胞系 16 小时。检查放线菌酮半衰期。用 C1(5 μM,16 小时)处理的 T47D 细胞显示 G1 期增加 (p < 0.0001) 和 S 期减少 (p < 0.0001),与 p27 蛋白诱导相关。相反,MCF-7 细胞对 C1 的反应是 G1 期显着减少(35%,p < 0.0001)和 G2-M 期增加(43%,p < 0.0001)。这种 G1 减少和 G2/M 停滞与 C1 剂量相关(图 5C 右;分别为 p < 0.001 和 p < 0.01),并与 p27 蛋白水平增加相关(图 5E 左,S4A 右上)。
1. 10 μM的SKPin C1可显著抑制多发性骨髓瘤(MM)U266和RPMI 8226细胞的活力,且抑制作用随剂量增加而增强;50 μM的SKPin C1在12小时内仅轻微降低正常B淋巴细胞的活力。与正常B淋巴细胞相比,U266和RPMI 8226细胞中Skp2表达水平更高,p27表达水平更低。SKPin C1处理或Skp2敲低可通过阻止p27泛素化来提高U266和RPMI 8226细胞中p27蛋白水平,进而减慢细胞周期、抑制细胞增殖并诱导细胞凋亡(通过MTT、EdU染色、细胞周期检测、蛋白质免疫印迹、免疫沉淀实验验证)。[1] 2. C1(Skp2抑制剂)在体外筛选(悬尾实验、强迫游泳实验、社交互动实验)中,于5 mg/kg和10 mg/kg剂量下(1 mg/kg剂量无此效果)对雌雄小鼠均表现出抗抑郁样作用;5 mg/kg C1以24小时内三次给药方式(测试前24、5、1小时)可在无应激小鼠中诱导抗抑郁样作用,而急性给药(测试前1小时)无此效果;C1与氟西汀联合给药对无应激小鼠的抑郁样行为产生累加效应,且C1对小鼠自主活动能力无影响。[2] 3. C1在体外可选择性抑制Skp2介导的p27泛素化,其机制为结合Skp2-Cks1结合界面(由Skp2-R294、Skp2-Y346、Cks1-R44、Cks1-Q52构成)并减少p27与Skp2的结合;10 μM C1可提高转移性黑色素瘤细胞系(501 Mel、SK-MEL-147、SK-MEL-173)中p27蛋白水平及半衰期,诱导细胞周期阻滞(501 Mel细胞阻滞于G1期,MCF-7细胞阻滞于G2/M期,T47D细胞阻滞于G1期),且对E2-Ubc3/E2-Ubc5泛素转移、CyclinE/CDK2介导的p27磷酸化、非靶向E3连接酶(MDM2、SCF-βTrCP)无抑制作用。[3] 4. C1可提高子宫内膜癌ECC-1细胞和原代子宫内膜癌(ECA)细胞中p27蛋白水平;与特异性提高核内p27的C2/C20不同,C1可同时提高ECC-1细胞胞质和核内的p27水平;C1可抑制原代ECA细胞(III/III级和I/III级肿瘤)的增殖。[4] |
| 体内研究 (In Vivo) |
通过尾悬试验(TST)、强迫游泳试验(FST)和社交互动试验(SIT),Skp2抑制剂C1(5 mg/kg和10 mg/kg;24小时内3次:24、5和测试前 1 小时)显示了长期治疗后的小鼠模型中的抗抑郁样作用。 [2]
1. 5 mg/kg C1(Skp2抑制剂)长期处理可改善慢性社会挫败应激小鼠的抑郁样行为,表明其在应激条件下具有抗抑郁样作用。[2] |
| 酶活实验 |
1. 体外泛素化实验:通过体外转录翻译生成重组蛋白(野生型/突变型Skp2、Cks1、p27);实验体系包含UBE1、His-UbcH3/His-UbcH5c、泛素、ATP,以及50 μM C1或溶媒,30℃孵育60分钟;加入非变性样品缓冲液(含/不含β-巯基乙醇)终止反应,通过蛋白质免疫印迹检测p27多聚泛素化水平。[3]
2. 泛素充电实验:将UBE1(100 nM)、His-UbcH3(10 ng/μl)与泛素(2.5 μg/μl)、ATP(2 mM)、10 μM C1或溶媒混合,30℃孵育60分钟;通过蛋白质免疫印迹分析E2充电活性,证实C1无抑制作用。[3] 3. 差示扫描荧光法:将重组His-6Skp1-Skp2-Cks1(1.5 μM)与75 μM C1、非匹配化合物(UM-C1)或溶媒(0.5% DMSO)预孵育;以0.06℃/s的升温速率(20-85℃)检测荧光(激发波长465 nm,发射波长580 nm)并计算熔解温度(Tm),结果显示C1可改变Skp2-Cks1复合物的Tm值(C1处理组48.51℃ vs 溶媒组44.09℃,UM-C1组45.06℃)。[3] 4. 表面等离子体共振(SPR)实验:采用纯化的Skp2-Cks1复合物验证C1与该复合物的结合作用。[3] |
| 细胞实验 |
细胞系:THP-1、U266、RPMI 8226 和 B 淋巴细胞
浓度:0、5、10、25 和 50 μM 孵育时间:12 小时、24 小时、36 小时和 48 小时< br> 结果:10 μM 浓度下,U266 和 RPMI 8226 细胞活力显着降低 12 小时。 50 μM 浓度下,对 B 细胞活力没有明显影响。 THP-1 细胞活力在 12 小时内降低。 50 μM 12 小时。 1. 多发性骨髓瘤细胞实验:将正常B淋巴细胞、THP-1、U266、RPMI 8226细胞暴露于不同剂量的SKPin C1中48小时;不同时间点通过MTT法检测细胞活力;处理12小时后通过流式细胞术分析细胞周期;采用蛋白质免疫印迹检测Skp2、p27、剪切型caspase-3蛋白水平;通过免疫沉淀实验检测p27泛素化水平;对25 μM SKPin C1或Skp2 siRNA处理的U266/RPMI 8226细胞,采用EdU染色评估细胞增殖情况。[1] 2. 黑色素瘤细胞实验:将501 Mel、SK-MEL-147、SK-MEL-173细胞用10 μM C1或溶媒处理16小时;制备全细胞提取物,通过蛋白质免疫印迹检测p27、p21、Skp2、Skp1、Cul1水平;采用环己酰亚胺追踪实验(20 ng/ml环己酰亚胺)评估p27半衰期(C1处理后从1小时延长至>5小时);利用慢病毒四环素诱导型shRNA(shSkp2/shp27)敲低Skp2/p27,随后用10 μM C1处理16小时,通过流式细胞术分析细胞周期。[3] 3. 乳腺癌/前列腺癌细胞实验:将MCF-7、T47D(乳腺癌)和LNCaP(前列腺癌)细胞分别用5 μM(MCF-7/T47D)或10 μM(LNCaP)C1处理16小时;通过流式细胞术分析细胞周期(MCF-7细胞阻滞于G2/M期,T47D/LNCaP细胞阻滞于G1期);采用蛋白质免疫印迹检测p27、Skp2、Skp1水平。[3] 4. 子宫内膜癌细胞实验:将ECC-1细胞用C1 处理18小时;进行亚细胞组分分离(核/胞质),通过蛋白质免疫印迹检测p27水平(以α-微管蛋白/特异性蛋白1作为组分纯度对照);采用MTS实验评估C1对细胞增殖的抑制作用;利用超分辨荧光定位显微镜量化核内p27簇(密度无数值变化,簇面积增加1.8倍)。[4] 5. 原代ECA细胞实验:将原代ECA细胞(I/I级、III/III级、I/III级、II/III级肿瘤)用C1 处理;通过蛋白质免疫印迹检测总细胞裂解液/核-胞质组分中的p27水平;采用MTS实验评估增殖抑制作用。[4] |
| 动物实验 |
1. 小鼠抗抑郁作用试验:雄性/雌性小鼠(品系未指定)经未指定途径分别给予1、5、10 mg/kg剂量的C1;试验包括急性给药(试验前1小时)和慢性给药(治疗8天);对于未经历过应激的小鼠,在24小时内进行三次给药(试验前24、5、1小时);对于经历过慢性社会挫败应激的小鼠,长期给予5 mg/kg C1治疗,以评估抑郁样行为(悬尾试验、强迫游泳试验、社交互动试验);测量运动活性以排除非特异性效应。[2] 2. 小鼠子宫内膜上皮细胞增殖试验:切除卵巢的C57Bl/6小鼠肌内注射100 ng 17-β-雌二醇(E2)2天,随后休息2天;小鼠连续两天腹腔注射C1(剂量未报告)或载体,24小时后处死;对子宫内膜组织进行免疫组织化学(IHC)检测,以检测p27(核)和PH3(增殖标志物)的水平(该检测中未报告C1的数据,仅报告了C5/C2的数据)。[4] |
| 毒性/毒理 (Toxicokinetics/TK) |
1. 50 μM SKPin C1 在 12 小时内仅轻微降低正常 B 淋巴细胞的活力,表明其对正常细胞的体外毒性较低。[1]
2. C1(5、10 mg/kg)对小鼠的运动活性无影响,表明其无与运动功能相关的急性毒性作用;(LD50、肝毒性、肾毒性、血浆蛋白结合率)已报道。[2] 3. C1(10 μM)对 501 Mel、MCF-7、T47D 和 LNCaP 细胞无细胞毒性。[3] 4. C1 对 ECC-1 细胞无细胞毒性(未观察到细胞活力降低,PARP1/caspase-3 裂解未检测到细胞凋亡)。[4] |
| 参考文献 | |
| 其他信息 |
1. SKPin C1 是一种 Skp2 抑制剂,此前已证实其对转移性黑色素瘤细胞具有抑制作用;它通过稳定 p27(阻止泛素化)减缓细胞周期并诱导细胞凋亡,从而有效抑制多发性骨髓瘤的异常增殖和永生化。[1]
2. C1 是一种特异性 Skp2 抑制剂;抑制 Skp2 可能是治疗抑郁症的潜在策略,Skp2 也可能是新型抗抑郁药研发的靶点;C1 与氟西汀联用可增强其缓解抑郁样行为的效果。[2] 3. C1 是一种小分子抑制剂,通过计算机虚拟配体筛选 (VLS) 鉴定,靶向 Skp2-Cks1 界面(由 Skp2-R294、Skp2-Y346、Cks1-R44 和 Cks1-Q52 形成的口袋)。它通过竞争性抑制 Skp2-p27 相互作用来阻断 Skp2 介导的 p27 降解,其细胞周期阻滞效应取决于细胞类型(黑色素瘤/前列腺癌/子宫内膜癌细胞为 G1 期,乳腺癌细胞为 G2/M 期)。[3] 4. C1 是一种 Skp2/Cks1 E3 连接酶抑制剂 (Skp2E3LI),可稳定子宫内膜癌细胞中的 p27(核内和胞质内),抑制 E2 诱导的 p27 降解和细胞增殖;它对以 SCF-Skp2/Cks1 介导的核内 p27 破坏为特征的子宫内膜癌具有潜在的治疗价值,并且优于一般的蛋白酶体抑制剂。[4] |
| 分子式 |
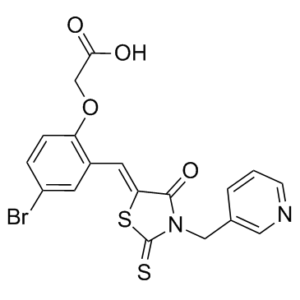
C18H13BRN2O4S2
|
|
|---|---|---|
| 分子量 |
465.34
|
|
| 精确质量 |
463.95
|
|
| 元素分析 |
C, 46.46; H, 2.82; Br, 17.17; N, 6.02; O, 13.75; S, 13.78
|
|
| CAS号 |
432001-69-9
|
|
| 相关CAS号 |
|
|
| PubChem CID |
5733396
|
|
| 外观&性状 |
White to yellow solid powder
|
|
| LogP |
3.646
|
|
| tPSA |
137.12
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
7
|
|
| 可旋转键数目(RBC) |
6
|
|
| 重原子数目 |
27
|
|
| 分子复杂度/Complexity |
631
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
S=C(S/1)N(CC2=CN=CC=C2)C(C1=C\C(C=C(Br)C=C3)=C3OCC(O)=O)=O
|
|
| InChi Key |
O=C(O)COC1=CC=C(Br)C=C1/C=C(SC(N2CC3=CC=CN=C3)=S)/C2=O
|
|
| InChi Code |
InChI=1S/C18H13BrN2O4S2/c19-13-3-4-14(25-10-16(22)23)12(6-13)7-15-17(24)21(18(26)27-15)9-11-2-1-5-20-8-11/h1-8H,9-10H2,(H,22,23)/b15-7-
|
|
| 化学名 |
2-[4-Bromo-2-[[4-oxo-3-(3-pyridinylmethyl)-2-thioxo-5-thiazolidinylidene]methyl]phenoxy]acetic acid
|
|
| 别名 |
MDK-1699; MDK 1699; MDK1699; Skp2 inhibitor C1 and SKPin C1.
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO :20.83~ 93 mg/mL ( 44.76~199.85 mM )
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (4.47 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: 5% DMSO + Corn oil: 1.2mg/ml 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1490 mL | 10.7448 mL | 21.4897 mL | |
| 5 mM | 0.4298 mL | 2.1490 mL | 4.2979 mL | |
| 10 mM | 0.2149 mL | 1.0745 mL | 2.1490 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
") In silicoandin vitroscreens for Skp2 ligase inhibitors.Chem Biol. 2012 Dec 21; 19(12): 1515–1524. |
|---|
") Structure-function approach to identify key contacts.Chem Biol. 2012 Dec 21; 19(12): 1515–1524. |
") Binding of inhibitors disrupts the Skp2-p27 interaction.Chem Biol. 2012 Dec 21; 19(12): 1515–1524. |
") Inhibitors induces p27 protein in melanoma cells.Chem Biol. 2012 Dec 21; 19(12): 1515–1524. |
|---|
") Skp2 ligase inhibitors induce cell cycle changes.Chem Biol. 2012 Dec 21; 19(12): 1515–1524. |
") Skp2E3LIs increase nuclear p27 in the ECA cell line, ECC-1.Endocrinology.2013 Nov;154(11):4030-45. |
") Skp2E3LI, C2, increases nuclear p27 in ECC-1 cells determined by super resolution fluorescent microscopy and quantitation.Endocrinology.2013 Nov;154(11):4030-45. |
|---|
") The Skp2E3LIs, C5, C2, and C20, inhibit cell proliferation, block cells in G1, are not cytotoxic, and do not induce apoptosis in ECC-1 cells.Endocrinology.2013 Nov;154(11):4030-45. |
") The Skp2E3LI, C2 increases nuclear p27 while decreasing p27 in the cytoplasm over time; C2 and C20 increase p27 half-life over controls in ECC-1 cells.Endocrinology.2013 Nov;154(11):4030-45. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
