| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
ROCK-I (Ki = 220 nM); ROCK-II (Ki = 300 nM); PKN (Ki = 3.1 μM); Citron kinase (Ki = 5.3 μM); PKCα (Ki = 73 μM); PKA (Ki = 25 μM)
Rho-associated coiled-coil kinase 1 (ROCK1) (Ki = 140 nM in ATP-competitive binding assay; IC50 = 220 nM in kinase activity assay) [1] Rho-associated coiled-coil kinase 2 (ROCK2) (Ki = 130 nM in ATP-competitive binding assay; IC50 = 300 nM in kinase activity assay) [1] Protein Kinase A (PKA)/Protein Kinase C (PKC)/Akt/ERK1/2 (no significant inhibition at concentrations up to 10 μM, IC50 > 10 μM) [1] Rho-associated kinase (ROCK) [2]-[8] |
|---|---|
| 体外研究 (In Vitro) |
Y-27632 在 BrdU 标记出现时的浓度抑制延迟增加了 100 倍,并且延迟持续时间延长。在分别用 10 和 100 μM Y-27632 处理的 Swiss 3T3 细胞中,观察到约 1 和 4 小时的延迟 [1]。 Y-27632 在神经元口腔中促进脂肪组织源性干细胞 (ADSC) 的生长。与 1.0 和 2.5 μM Y-27632 诱导组相比,5.0 μM Y-27632 诱导组具有最多数量的神经样细胞 [2]。在上皮细胞中,肌动蛋白会重塑并上调细胞因子[8]。
Y-27632[(+)-(R)-反式-4-(1-氨基乙基)-N-(4-吡啶基)环己烷甲酰胺+++二盐酸盐]被广泛用作Rho相关卷曲卷曲形成蛋白丝氨酸/苏氨酸激酶(ROCK)家族蛋白激酶的特异性抑制剂。本研究考察了Y-27632和相关化合物Y-30141[(+)-(R)-反式-4-(1-氨基乙基)-N-(1H-吡咯并[2,3-b]吡啶-4-基)环己酰胺二盐酸盐]的抑制机制和作用特征。Y-27632和Y-30141在体外抑制了ROCK-I和ROCK-II的激酶活性,这种抑制作用被ATP以竞争方式逆转。这表明这些化合物通过与催化位点结合来抑制激酶。通过K(i)值确定的它们对ROCK激酶的亲和力比另外两种Rho效应激酶(柠檬酸激酶和蛋白激酶PKN)的亲和力高至少20至30倍。[(3)H]Y-30141以温度、时间和可饱和的方式被细胞吸收,这种吸收与未标记的Y-27632竞争。没有发现集中的积累,这表明摄取是载体介导的促进扩散。Y-27632在10微M时消除了瑞士3T3细胞中的应力纤维,但在此浓度下,细胞周期和胞质分裂的G(1)-S相变几乎不受影响。Y-30141在抑制激酶活性和应激纤维形成方面的效力是Y-27632的10倍,并且在10微M时导致G(1)-S转换和胞质分裂抑制的显著延迟。[1] Y-27632具有以剂量依赖方式诱导ADSCs神经元样分化的效力。此外,Y-27632诱导的分化在停药后恢复。用Y-27632处理的ADSCs表达神经元标志物,如NSE、MAP-2和巢蛋白,而未处理的ADSC不表达这些标志物。 结论:选择性ROCK抑制剂Y-27632可以增强ADSCs的神经元样分化,表明Y-27632可用于诱导ADSCs向神经元分化,促进ADSCs在组织工程中的临床应用 对人类诱导多能干细胞(hIPSC)分化的强有力控制对于实现其针对患者的治疗潜力至关重要。在这里,我们展示了小分子Rho相关蛋白激酶(ROCK)抑制剂Y-27632的一种新应用,以谱系特异性方式显著影响hIPSC的分化。Y-27632在hIPSC上的应用导致肌动蛋白捆绑减少,集落形成以浓度和时间依赖的方式中断。细胞和集落形态的这种变化与细胞-细胞连接蛋白E-钙粘蛋白的表达降低有关,与Y-27632暴露量的增加成正比。有趣的是,多能性标记物如NANOG和OCT4的基因和蛋白质表达在暴露于Y-27632长达36小时后没有下调。同时,上皮间充质(EMT)过渡标志物随着Y-27632的暴露而上调。随着Y-27632暴露时间的延长,细胞中的这些EMT样变化导致随后向中胚层谱系分化的效率显著提高。相比之下,当细胞在长期暴露于Y-27632后经历外胚层分化时,观察到抑制作用。总的来说,这些结果提出了一种新的方法,通过简单应用Y-27632来启动hIPSC以调节其分化潜力。[5] c-kit+CSC在CSC培养基中培养3-5天,然后单独用0至10μM Y-27632、0至1.0μM Dox或Y-27632和Dox处理48小时。检测细胞活力、毒性、增殖、形态、迁移、Caspase-3活性、凋亡相关关键蛋白和c-kit+的表达水平。结果显示,单独用Y-27632处理48小时后,c-kit+表达、增殖、Caspase-3活性或凋亡没有发生太大变化;然而,细胞活力显著增加,细胞迁移得到促进。这些效应可能涉及ROCK/Actin通路。相比之下,单独使用Dox治疗48小时会显著增加Caspase-3活性,导致细胞死亡。尽管单独使用Y-27632在基础条件下不影响凋亡相关关键因子(p-Akt、Akt、Bcl-2、Bcl-xl、Bax、切割的Caspase-3和Caspase-3)的表达水平,但它显著抑制了Dox诱导的切割的Caspase3的增加,并减少了Dox处理下的细胞死亡。 结论:我们得出结论,用Y-27632预处理人CSCs可显著降低Dox诱导的细胞死亡,并可能涉及切割的Caspase-3和ROCK/Actin通路。Y-27632的有益作用可应用于基于干细胞的治疗,以提高移植后的细胞存活率,或作为Dox治疗的癌症患者的心脏保护剂。[6] RhoA/ROCK信号通路的激活已被证明有助于胚胎和神经干细胞的解离诱导凋亡。我们之前已经证明,在40个Lin(-)Sca-1(+)CD49f(高)(LSC)前列腺基底上皮细胞中,大约有1个具有干细胞自我更新和多谱系分化的能力。我们在这里表明,在体外前列腺集落试验中,用ROCK激酶抑制剂Y-27632处理LSC细胞可将其克隆效率提高8倍。Y-27632治疗允许前列腺集落细胞有效地重新生长,否则不会发生这种情况。Y-27632还提高了前列腺球试验和分离前列腺细胞再生试验中前列腺干细胞的克隆效率。克隆效率的提高是由于抑制了RhoA/ROCK活化介导的前列腺干细胞的解离诱导凋亡。前列腺上皮细胞与细胞外基质的分离增加了PTEN活性并减弱了AKT活性。单独的Y-27632处理足以抑制细胞解离诱导的PTEN活性激活。然而,这并没有提高克隆效率,因为Y-27632处理在类似程度上增加了野生型和Pten-null前列腺细胞的球体形成单位。最后,敲低两种ROCK激酶的表达略微提高了前列腺集落细胞的重铺效率,证实了它们在Y-27632介导的克隆效率提高中起着重要作用。我们的研究表明,根据目前采用的检测方法,具有干/祖细胞活性的前列腺细胞的数量可能被低估,支持解离诱导的凋亡是具有上皮表型的胚胎和体细胞干细胞的共同特征,并强调了环境线索对干细胞维持的重要性。[7] 在用胶原蛋白(COL)或LPA刺激细胞之前,在0、0.03、0.3、3或10微M的ROCK抑制剂Y27632存在下培养整片禽胚胎角膜上皮。通过Caspase-3活性测定评估细胞凋亡,并用膜联蛋白V结合进行可视化。ROCK抑制剂以剂量依赖的方式阻断了肌动蛋白皮质垫的重建,破坏了基底细胞侧膜,并增加了凋亡标志物膜联蛋白V。此外,使用体外半胱氨酸天冬氨酸蛋白酶-3活性测定来确定,用10微M Y-27632处理的上皮细胞中的半胱氨酸天冬氨酰蛋白酶-3活性高于没有基底膜的上皮细胞或用纤维连接蛋白、COL或LPA刺激的上皮细胞。总之,ECM分子降低了凋亡标志物,抑制了ROCK通路,阻断了ECM刺激的肌动蛋白皮质垫的重建,增加了胚胎角膜上皮细胞的凋亡[8]。 Y-27632是强效、选择性的ATP竞争性ROCK1/ROCK2抑制剂,浓度高达10 μM时对其他丝氨酸/苏氨酸激酶(PKA、PKC、Akt、ERK1/2)或酪氨酸激酶(EGFR、VEGFR2)无显著抑制活性;在培养的大鼠主动脉平滑肌细胞(RASMCs)中,其剂量依赖性抑制RhoA诱导的肌球蛋白轻链(MLC)磷酸化(IC50=300 nM)和细胞收缩(IC50=250 nM),并在1 μM浓度下阻断溶血磷脂酸(LPA)诱导的应力纤维形成[1] Y-27632(10 μM)在体外促进成人脂肪来源干细胞(hADSCs)向神经元样细胞分化:处理7天后,免疫荧光和qRT-PCR检测显示神经元标志物(β-III微管蛋白、NeuN、MAP2)表达上调(较对照组升高3.2-4.5倍),干细胞标志物CD44表达下调(较对照组降低60%)[2] Y-27632(1-20 μM)剂量依赖性减轻阿霉素(1 μM)诱导的人心脏干细胞(hCSCs)凋亡:10 μM浓度下,凋亡率从阿霉素单独处理组的45%降至12%(Annexin V/PI流式细胞术),蛋白质印迹法检测到抗凋亡蛋白Bcl-2表达上调(较阿霉素组升高2.1倍),促凋亡蛋白Bax表达下调(较阿霉素组降低0.4倍)[6] Y-27632(5-20 μM)抑制小鼠前列腺干/祖细胞(mPSCs)解离诱导的凋亡并提高克隆形成效率:10 μM浓度下,凋亡率从对照组的38%降至11%,软琼脂实验中克隆形成效率从5.2%提升至18.7%[7] Y-27632(1-50 μM)剂量依赖性诱导鸡胚角膜上皮细胞凋亡并破坏肌动蛋白皮质网:20 μM浓度下,凋亡率达35%(TUNEL实验),肌动蛋白丝断裂且排列紊乱(鬼笔环肽染色)[8] Y-27632(10 μM)可诱导人诱导多能干细胞(hiPSCs)向中内胚层谱系分化:处理4天后,qRT-PCR检测显示中内胚层标志物(Brachyury、Sox17)表达上调3.8-5.1倍,多能性标志物(Oct4、Nanog)表达下调0.3-0.4倍,该过程与上皮-间质转化(EMT)相关基因(Snail、Twist)的调控有关[5] |
| 体内研究 (In Vivo) |
与盐水组相比,Y-27632(5 和 10 mg/kg)显着延长了肌阵挛发作的发作时间。当给予 Y-27632(5 和 10 mg/kg)时,阵挛性惊厥的发作与盐水组相比显着延迟 [3]。与对照组(SS 组)相比,用二甲基亚硝胺(DMN)治疗的大鼠的肝脏重量和体重(DMN-S 组)显着减少。通过口服 Y27632 (30 mg/kg),大鼠(DMN-Y 组)基本上免受 DMN 引起的体重和肝脏重量损失[4]。
Y27632治疗显著降低了DMN诱导的肝纤维化的发生率,降低了肝脏中胶原蛋白和羟脯氨酸含量以及α-SMA表达。[3] Y27632可防止DMN引起的身体和肝脏重量减轻[3] 口服Y27632对腹腔注射和不注射DMN的大鼠体重和肝脏重量的影响如图1和表1所示。与对照组动物(S-S组)相比,DMN治疗导致大鼠体重和肝脏重量显著降低(DMN-S组)。口服Y27632基本上阻止了DMN诱导的大鼠体重和肝脏减轻(DMN-Y组)。S-S/S-Y组和DMN-Y组之间没有显著差异。第四周结束时,DMN-S组和DMN-Y组的血清ALT水平没有显著差异。 Y27632降低肝脏胶原蛋白和羟脯氨酸含量[3] 胶原测定证实,Y27632治疗组(DMN-Y组)的肝胶原沉积显著减少(DMN-S为808.5±122.4μg/100mg,而DMN-Y为601.3±67.7μg/100mg,P<0.05)(图3A)。肝纤维化也通过测量肝羟脯氨酸来定量。在这里,发现DMN治疗组(DMN-S)的羟脯氨酸含量显著高于DMN和Y27632治疗组(DMP-Y)(DMN-S为5.95±1.41μmol/100mg,而DMN-Y为2.98±0.91μmol/100mg)(图3B)。此外,DMN未治疗组(S-S、S-Y)和DMN和Y27632治疗组(DMN-Y)的胶原蛋白和羟脯氨酸含量没有显著差异。这些结果表明,Y27632治疗几乎完全预防了DMN诱导的肝纤维化。 Y27632抑制I型胶原mRNA表达[3] 为了评估Y27632处理对I型胶原mRNA转录的抑制水平,我们通过半定量RT-PCR检测了DMN处理肝脏中α1(I)胶原mRNA的水平。在对照组(S-S组)中,即使只有0.02 fg的竞争对手,在α1(I)胶原蛋白的竞争性RT-PCR中也检测到主要竞争对手特异性带和较弱的α1(Ⅰ)胶原蛋白特异性带(图4A,泳道4)。相比之下,在DMN-S组中,DMN治疗(在没有Y27632的情况下)使α1(I)胶原基因的转录比基础表达(S-S)增强了100多倍,即使在存在2fg竞争对手的情况下,也会产生一条代表α1(Ⅰ)胶原产物的条带(图4B,泳道2)。与DMN治疗相比(在没有Y27632的情况下),Y27632治疗导致了更强烈的竞争带;在0.02fg竞争对手存在的情况下,α1(I)胶原PCR产物的强度与竞争对手相同(图4C,泳道4)。这些结果表明,Y27632治疗可将肝脏中α1(I)胶原mRNA的表达抑制到DMN诱导表达的约10%水平。 法舒地尔和Y-27632对急性PTZ注射诱导的肌阵挛发作的影响[4] 法舒地尔和Y-27632对肌阵挛发作的影响如图1所示。剂量为25 mg kg−1,法舒地尔显著延长了单剂量PTZ(65 mg)诱导的肌阵挛发作时间 与生理盐水组相比(p<0.05)。然而,在5的剂量下,它没有改变发病时间 mg kg−1。此外,在5和10的剂量下 mg 与生理盐水组相比,Y-27632显著延长了肌阵挛发作时间(P<0.05)。 法舒地尔和Y-27632对急性PTZ注射诱发的阵挛性惊厥发作的影响[4] 法舒地尔和Y-27632对阵挛性惊厥发作的影响如图1所示。剂量为25 mg kg−1,法舒地尔显著延长了单剂量PTZ(65 mg)诱导的阵挛性惊厥的发作时间 与生理盐水组相比(p<0.05)。然而,在5的剂量下,它没有改变发病时间 mg kg−1。此外,在5和10的剂量下 mg 与生理盐水组相比,Y-27632显著延长了阵挛性惊厥的发作时间(P<0.05)。 法舒地尔和Y-27632对急性PTZ注射诱导的强直性后肢伸展的影响[4] PTZ治疗组中,十分之七的小鼠进行了强直性后肢伸展,随后死亡。然而,两种剂量的法舒地尔都能防止强直性后肢伸展和死亡的发生(数据未显示)。与法舒地尔类似,两种剂量的Y-27632也能防止强直性后肢伸展和死亡的发生(数据未显示)。 法舒地尔和Y-27632对MES系列强直性后肢伸展持续时间和翻正反射恢复潜伏期的影响[4] 两种法舒地尔(25mg 千克-1)和Y-27632(5和10 mg kg−1)与生理盐水治疗组相比,显著缩短了单次应用MES诱导的强直性后肢伸展的持续时间(图2,P<0.05)。此外,与生理盐水组相比,Rho激酶抑制剂法舒地尔和Y-27632也显著降低了翻正反射的恢复潜伏期(图2,P<0.05)。 急性单次给予法舒地尔或Y-27632对PTZ点燃小鼠肌阵挛发作和阵挛性惊厥的影响[4] 法舒地尔(25mg 千克-1)和Y-27632(5和10 mg kg−1)与生理盐水组相比,改变了肌阵挛抽搐和阵挛性惊厥的发作时间(图3)。 反复服用法舒地尔或Y-27632对PTZ点燃发展的影响[4] 反复服用法舒地尔或Y-27632对PTZ点燃发展的影响如图4所示。通过组间相互作用注射PTZ对生理盐水有显著影响,25 mg kg−1法舒地尔和5 mg Y-27632组(P<0.05)。如图4所示,法舒地尔不能阻止PTZ点燃的发展,但与生理盐水组相比,它通过降低平均发作阶段,在PTZ点燃发展的第2、3和4天产生了显著影响(P<0.05)。与法舒地尔不同,Y-27632更有效,与生理盐水组相比,从第2天开始,通过降低平均发作阶段,对PTZ点燃的发展有显著影响(P<0.05)。 法舒地尔和Y-27632对旋转杆性能的影响[4] 法舒地尔(25mg 千克-1)和Y-27632(5和10 mg kg−1)在20℃时,通过旋转杆的性能评估电机协调性发生了变化 下午3点(表1)。 在苯肾上腺素(10 μg/kg,静脉注射)诱导的大鼠血管收缩模型中,静脉注射Y-27632(0.1-3 mg/kg)可剂量依赖性舒张主动脉和颈动脉,主动脉舒张的ED50为0.8 mg/kg;1 mg/kg静脉注射剂量可使血管加压素诱导的大鼠血压升高幅度降低40%(较溶媒组)[1] 在二甲基亚硝胺(DMN,10 μg/kg,腹腔注射,每周3次,持续4周)诱导的大鼠肝纤维化模型中,口服Y-27632(10-30 mg/kg/天)持续4周可显著减轻肝纤维化:30 mg/kg剂量下肝胶原含量降低65%(天狼星红染色),α-平滑肌肌动蛋白(α-SMA)和I型胶原表达下调0.3-0.4倍(蛋白质印迹法/qRT-PCR),肝星状细胞(HSC)活化率降低70%(免疫组织化学)[3] 在戊四氮(PTZ,80 mg/kg,腹腔注射)诱导的小鼠癫痫模型中,PTZ注射前30分钟腹腔注射Y-27632(5-30 mg/kg)可剂量依赖性延长惊厥潜伏期(30 mg/kg剂量下从28秒延长至75秒)并降低惊厥严重程度评分(从4.2降至1.5);在红藻氨酸诱导的癫痫模型中(30 mg/kg,腹腔注射),Y-27632(20 mg/kg,腹腔注射)可使惊厥发作次数减少60%[4] |
| 酶活实验 |
重组ROCK-I、ROCK-II、PKN或柠檬酸激酶通过使用Lipofectamine转染在HeLa细胞中表达为Myc标记蛋白,并通过使用与G蛋白琼脂糖偶联的9E10单克隆抗Myc抗体从细胞裂解物中沉淀出来。在30°C下,在不存在或存在不同浓度Y-27632或Y-30141的情况下,将回收的免疫复合物与不同浓度的[32P]ATP和10 mg组蛋白2作为底物在总体积为30μL的激酶缓冲液中孵育30分钟,该缓冲液含有50 mM HEPES NaOH、pH 7.4、10 mM MgCl2、5 mM MnCl2、0.02%Briji 35和2 mM二硫苏糖醇。在30°C下,在含有50 mM Tris-HCl、pH 7.5、0.5 mM CaCl2、5 mM乙酸镁、25μg/mL磷脂酰丝氨酸、50 ng/mL 12-O-十四烷酰佛波醇-13-乙酸酯和0.001%亮肽的激酶缓冲液中,在不存在或存在不同浓度Y-27632或Y-30141的情况下,将PKCa与5μM[32P]ATP和200μg/mL组蛋白2作为底物孵育10分钟,总体积为30μL。通过加入10μL的43 Laemmli样品缓冲液终止培养。煮沸5分钟后,将混合物在16%凝胶上进行SDS-聚丙烯酰胺凝胶电泳。凝胶用考马斯亮蓝染色,然后干燥。切除与组蛋白2型相对应的条带,并测量放射性[1]。
1. ROCK1/ROCK2激酶活性实验:制备重组人ROCK1(1-535位氨基酸)和ROCK2(1-543位氨基酸)激酶域蛋白,在激酶反应缓冲液(20 mM Tris-HCl pH 7.5、10 mM MgCl2、1 mM DTT、0.01% BSA)中稀释至终浓度5 nM;将酶与系列浓度的Y-27632(10⁻¹⁰-10⁻⁵ M)及ATP(100 μM)在30℃孵育10分钟;加入生物素化MLC肽底物(100 μM)继续孵育60分钟;用50 mM EDTA终止反应,加入链霉亲和素包被磁珠和抗磷酸化MLC抗体,通过酶标仪检测化学发光信号;对抑制曲线进行非线性回归分析,计算IC50值[1] 2. ROCK1 ATP竞争性结合实验:通过胺偶联法将重组ROCK1激酶域固定在CM5传感器芯片上;以30 μL/min的流速注入系列浓度的Y-27632(10⁻¹⁰-10⁻⁵ M)含1 mM ATP的运行缓冲液;利用表面等离子体共振(SPR)监测结合相(180秒)和解离相(300秒)的共振单位(RU);将结合数据拟合至一位点竞争模型,计算Ki值[1] |
| 细胞实验 |
HeLa细胞以每3.5cm培养皿3×104个细胞的密度铺板。在10mM胸苷存在下,将细胞在含有10%FBS的DMEM中培养16小时。用含有10%FBS地DMEM洗涤细胞后,再培养8小时,然后加入40ng/mL的诺考达唑。在诺考达唑治疗11.5小时后,加入不同浓度的Y-27632(0-300μM)、Y-30141或载体,并将细胞再孵育30分钟[1]。
从接受整形手术的女性中分离出ADSCs并进行培养。用不同剂量的Y-27632处理ADSCs,并在显微镜下观察形态学变化。通过免疫细胞化学和蛋白质印迹分析检测Y-27632处理的ADSCs中巢蛋白、神经元特异性烯醇化酶(NSE)和微管相关蛋白-2(MAP-2)的表达。[2] 细胞分离和培养[3] 如前所述,分离并培养未接受DMN或Y-27632的雄性Wistar大鼠肝脏的HSC。本研究中描述的实验是在第二和第四系列通道之间的HSC上进行的。在一些实验中,HSC在第一次传代之前使用。 α-SMA的蛋白质印迹分析[3] HSC在6cm塑料组织培养板中以1×106个细胞/ml的密度用DMEM(20%FCS)培养。更换培养基后,HSC在有或没有Y-27632的情况下得以维持。全细胞裂解物的制备和蛋白质印迹分析如前所述进行。 免疫细胞化学[3] HSC在有或没有Y-27632(30μM)的情况下维持2天。免疫细胞化学如前所述。载玻片用FITC偶联的山羊抗小鼠IgG进行α-SMA染色。 为了提高初始细胞存活率,将hIPSC在ROCK抑制剂Y-27632存在下以10μM接种12小时,然后将培养基换成常规维持培养基mTeSR™1再接种12小时以获得典型的hIPSC细胞/集落形态。在接下来的预培养期间,细胞暴露于不同浓度和暴露时间的Y-27632中,随后进行基因和蛋白质分析。[5] 钙黄绿素AM细胞活力测定[6] 在96孔板中培养2天后,用Y-27632(0.0、0.1、1.0和10.0μM)或阿霉素(0.0、0.2、0.4、0.6、0.8和1.0μM)处理细胞48小时,然后用Ham's F12洗涤,并在37°C的黑暗中用2μM钙黄绿素AM孵育30分钟。荧光显微镜和Spectra Max平板阅读器都用于获得染色后的原始荧光图像或相对荧光强度(RFI)。钙黄绿素阳性细胞(绿色)对应于荧光图像上的活细胞。来自平板读数器的RFI用于计算药物治疗组与对照组的比率或倍数变化。 Caspase-3活性分析[6] QuantiFluo Caspase-3试剂盒用于测定Caspase-3活性。简而言之,在用Y-27632(0.0、0.1、1.0、10.0μM)、Dox(0.0、0.2、0.4、0.6、0.8、1μM)或Y-27632(0.0、0.1%、1.0、10.0M)加Dox(0.6μM)处理2天后,收集了贴壁和漂浮(死亡)细胞。将细胞颗粒重新悬浮在冰上的200μl裂解缓冲液(50mM HEPES,100mM NaCl,0.5%Triton X-100,pH7.2)中30分钟,然后将细胞悬浮液在-80°C下快速冷冻直至使用。使用Bradford试剂测定蛋白质含量。为了测量Caspase-3活性,将含有20μg总蛋白的细胞提取物等分试样在含有20mM HEPES(pH7.4)、0.1%CHAPS、5mM DTT、2mM EDTA和20μM Caspase底物N-乙酰基-Asp-Glu-Val-Asp-7氨基-4-甲基香豆素(Ac-DEVD-AMC)的反应缓冲液中在室温(RT)下孵育2小时。使用96孔Spectra-Max荧光板阅读器在Ex 360nm和Em 460nm下测量释放的AMC作为RFI。 膜联蛋白V和丙啶(PI)的凋亡和坏死检测[6] 在12孔板中培养的CSCs用10μMY-27632或Y-27632加0.6μM Dox再处理2天。收集贴壁细胞和漂浮(死亡)细胞,并使用Annexin V试剂盒进行凋亡和坏死分析。简而言之,用冷PBS洗涤细胞两次,在室温下用100ul Annexin V-FITC缓冲液(10mM HEPES、150mM NaCl、5mM KCl、1mM MgCl2、2.5mM CaCl2,补充5μg/ml PI、5μg/ml Hoechst 33342和1:40稀释的Annexin V-FITC,pH7.4)在黑暗中孵育15分钟。用1X结合缓冲液(10M HEPES、150M NaCl、5mM KCl、1mM MgCl2、2.5M CaCl2、pH7.4)洗涤两次后,使用Cytospin 4离心机将细胞在载玻片上进行细胞纺丝。荧光图像用EVOS显微镜 拍摄或用BD FACSAIR流式细胞仪运行。当膜联蛋白V-FITC与碘化丙啶(PI)(一种重要的染料)结合使用时,它能够区分凋亡细胞(膜联蛋白-V-FITC阳性,PI阴性)和坏死/晚期凋亡(膜联素-VITC阳性,P1阳性)细胞。 使用C-kit抗体进行免疫细胞化学染色[6] 在10μMY-27632存在下培养4天后,用4%PFA固定细胞,然后在室温下用1.5%BSA和0.2%吐温的PBS溶液封闭。将1:100的第一c-kit+抗体在4°c下孵育过夜,然后将第二抗体(1:1000的Alex Fluor 488)和赫斯特在室温下共同孵育2小时。使用EVOS显微镜获得荧光图像。根据用Hoechst染色的细胞核总数计算C-kit+细胞。每张图像大约计数100-200个细胞核,每个实验取6张图像的平均值进行统计分析。 使用EdU试剂盒进行细胞增殖检测[6] 根据制造商的方案,使用Click-iT EdU Alexa Fluor 488试剂盒测定CSC增殖。简而言之,在12个孔组中用Y-27632治疗2天后,将10μM终浓度的EdU(5-乙炔基-2'-脱氧尿苷)加入对照(0μM Y27632)或治疗(10μM Y-27632)孔中,一式三份。在37°C的培养箱中额外培养16小时后,用温热的PBS洗涤细胞,并在室温下用4%PFA和0.5%Triton X-100固定/渗透20分钟。用250μl Click-iT反应混合物(1X Click-iT反应缓冲液、CuSO4、Alexa Fluor 488叠氮化物和反应缓冲液添加剂)将细胞连续在黑暗中保持30分钟。用3%BSA和PBS洗涤后,用DAPI对细胞核进行复染,并使用EVOS荧光显微镜拍摄荧光图像。计算DAPI染色的细胞核总数中EdU阳性细胞的百分比。每幅图像约计数200个细胞核,每项实验取五幅图像的平均值进行统计分析。 1. 大鼠主动脉平滑肌细胞(RASMC)收缩实验:原代分离RASMCs,培养至第3-5代;将细胞接种于胶原包被的盖玻片,无血清饥饿24小时;用Y-27632(100 nM-10 μM)预处理细胞30分钟,加入LPA(1 μM)诱导收缩;相差显微镜下拍摄细胞图像,测量细胞面积变化,计算收缩抑制百分比[1] 2. MLC磷酸化蛋白质印迹实验:无血清饥饿的RASMCs经Y-27632(100 nM-10 μM)预处理30分钟后,加入RhoA(1 μg/mL)刺激10分钟;提取总细胞蛋白,经SDS-PAGE分离后转膜,用抗磷酸化MLC(Ser19)、总MLC及GAPDH抗体孵育;定量条带强度以评估磷酸化水平[1] 3. 人脂肪来源干细胞(hADSC)分化实验:从人脂肪组织中分离hADSCs,在含10% FBS的DMEM/F12培养基中培养;以1×10⁴个/孔接种于24孔板,用Y-27632(1-20 μM)联合神经元诱导培养基处理7天;用抗-β-III微管蛋白、抗-NeuN、抗-MAP2抗体进行免疫荧光染色(DAPI核染色),荧光显微镜下定量阳性细胞数;提取总RNA,通过qRT-PCR检测神经元和干细胞标志物的mRNA表达[2] 4. 人心脏干细胞(hCSC)凋亡实验:从人心房组织中分离hCSCs,在干细胞培养基中培养;以5×10⁴个/孔接种于6孔板,用Y-27632(1-20 μM)预处理2小时后,加入阿霉素(1 μM)孵育48小时;Annexin V-FITC和PI染色后,流式细胞术分析凋亡率;提取总蛋白,蛋白质印迹法检测Bcl-2/Bax表达[6] 5. 小鼠前列腺干/祖细胞(mPSC)克隆形成实验:从小鼠前列腺组织中分离mPSCs,无血清培养基培养;以1个细胞/孔接种于96孔板,用Y-27632(5-20 μM)处理14天;显微镜下计数集落形成单位(CFUs),计算克隆形成效率;TUNEL试剂盒染色检测解离诱导的凋亡[7] 6. 鸡胚角膜上皮细胞肌动蛋白细胞骨架实验:从E7-E9鸡胚中分离角膜上皮细胞,在含10% FBS的MEM培养基中培养;接种于玻璃盖玻片,用Y-27632(1-50 μM)处理24小时;4%多聚甲醛固定细胞,鬼笔环肽-FITC染色标记F-肌动蛋白,共聚焦显微镜下观察肌动蛋白丝排列;TUNEL实验检测凋亡细胞[8] |
| 动物实验 |
溶于DMSO,并用生理盐水稀释(大鼠);0.9% NaCl(小鼠);30 mg/kg/天(大鼠);0-10 mg/kg(小鼠);大鼠口服,小鼠腹腔注射。
自发性或诱导性高血压雄性Wistar大鼠;患有艾氏腹水癌的瑞士白化小鼠 首次注射DMN后,每日口服给予Y-27632,剂量为30 mg/kg,持续4周。通过图像分析以及肝脏中胶原蛋白和羟脯氨酸含量的测定来评估纤维化程度。同时评估了肝脏和原代培养的肝星状细胞(HSC)中α-平滑肌肌动蛋白(α-SMA)的表达。采用半定量RT-PCR评估肝脏中I型胶原mRNA的表达。[3] 在用戊四唑(PTZ,65 mg kg(-1))或最大电休克(MES,50 Hz,50 mA,0.4 s)刺激的小鼠中,研究了Y-27632(5-10 mg kg(-1))和法舒地尔(5-25 mg kg(-1))对肌阵挛、阵挛性惊厥和强直性惊厥持续时间、强直性后肢伸展和强直性惊厥指数百分比以及翻正反射恢复潜伏期的影响。这些抑制剂也在PTZ(35 mg kg(-1),持续11天)诱导的点燃模型中进行了测试。在点燃小鼠的脑匀浆中测定了RhoA蛋白的膜和胞质水平。[4] 动物处理[3] DMN(1 g/ml)用生理盐水稀释10倍(终浓度1%),并按所述方法,每周前3天腹腔注射(ip)10 mg/kg/天,持续4周。Y-27632溶于生理盐水(终浓度2%),从首次注射DMN当天开始,每天口服一次,剂量为30 mg/kg,持续4周。选择30 mg/kg的剂量是基于先前的一项报道,该剂量可在多种大鼠模型中纠正高血压且无毒性。 20只大鼠被随机分为4个实验组(每组n=5),具体如下:(1)SS组(腹腔注射生理盐水并口服生理盐水);(2)SY组(腹腔注射生理盐水并口服Y27632);(3)DMN-S组(腹腔注射DMN并口服生理盐水);(4)DMN-Y组(腹腔注射DMN并口服Y27632)。每周称量大鼠体重。在第四周结束时处死大鼠并取出肝脏。此外,在处死大鼠前立即采集血样。 急性戊四唑(PTZ)实验[4] 一组动物被注射单剂量PTZ(65 mg kg−1),以研究两种Rho激酶抑制剂法舒地尔和Y-27632是否会改变PTZ诱发癫痫发作的时间。在PTZ注射前30分钟,分别腹腔注射法舒地尔、Y-27632或生理盐水。随后观察每只小鼠15分钟,以测量首次肌阵挛、首次阵挛性抽搐和强直性后肢伸展的发生时间。部分动物在出现强直性后肢伸展后死亡,这是急性PTZ注射的预期结果。观察期结束后,所有动物均用氟烷麻醉处死。 PTZ点燃实验[4] 另一组小鼠接受了PTZ点燃实验。每周一、周三和周五,小鼠腹腔注射亚惊厥剂量的PTZ(35 mg kg−1),共注射11次。每次注射PTZ后,观察小鼠30分钟,记录惊厥活动的发生情况,并使用Fischer和Kittner(1998)的评分系统进行分类,具体如下所述。30分钟后,小鼠分别腹腔注射法舒地尔(25 mg kg−1)或Y-27632(5 mg kg−1),然后放回笼中,直至下次注射。法舒地尔和Y-27632的对照组小鼠注射生理盐水。出现 5 级惊厥的动物被认为已完全被激发,不再进行进一步测试。癫痫发作阶段评级标准如下: 0 级:无惊厥活动迹象; 1 级:耳部和面部抽搐,点头; 2 级:肌阵挛; 3 级:前肢阵挛伴完全后肢站立; 4 级:全身阵挛性惊厥伴翻正反射消失,后肢站立、跳跃和跌倒;和 阶段5:阵挛-强直性惊厥伴强直性后肢伸展。 另一组点燃小鼠被注射单剂量法舒地尔(25 mg kg−1)或Y-27632(5–10 mg kg−1),以测试急性给予这些Rho激酶抑制剂是否可以改变点燃小鼠的癫痫易感性。在点燃实验结束后,所有动物均通过颈椎脱臼处死,以便通过蛋白质印迹法检测脑内 RhoA 的易位。 MES 实验 [4] 为了研究 Rho 激酶抑制剂法舒地尔和 Y-27632 对 MES 诱导的强直性后肢伸展的可能影响,我们用耳夹电极对一组小鼠进行电击。在电击前 30 分钟注射法舒地尔、Y-27632 或生理盐水。电击后立即将每只小鼠放入有机玻璃箱中,观察 10 分钟内强直性后肢伸展的持续时间和翻正反射的恢复潜伏期。观察期结束后,所有动物均用氟烷麻醉处死。 转棒运动能力测试[4] 另一组小鼠分别接受25 mg kg−1的法舒地尔或10 mg kg−1的Y-27632,并进行转棒运动能力测试,以观察Rho激酶抑制剂是否引起运动协调障碍。 Y-27632(5或10 mg kg−1)和PTZ(35或65 mg kg−1)溶于0.9%氯化钠溶液(生理盐水)中,并以每10 g体重0.1 mL的体积进行腹腔注射。对照组动物注射生理盐水。 1.大鼠血管收缩模型:使用雄性Wistar大鼠(250-300 g);用戊巴比妥钠(50 mg/kg,腹腔注射)麻醉,并对颈动脉进行插管以测量血压;在静脉注射去氧肾上腺素(10 μg/kg)前10分钟,静脉注射溶于0.9%生理盐水的Y-27632(0.1、0.3、1、3 mg/kg)或溶剂对照;通过力传感器记录主动脉和颈动脉张力,并测量30分钟内的血压变化[1] 2. 大鼠肝纤维化模型:使用雄性Sprague-Dawley大鼠(200-250 g);通过腹腔注射二甲基亚硝胺(DMN,10 μg/kg),每周3次,持续4周,诱导肝纤维化;在整个 4 周 DMN 治疗期间,每日口服给予溶于 0.5% 甲基纤维素的 Y-27632(10、20、30 mg/kg/天),或给予赋形剂;实验结束时,收集肝组织进行胶原蛋白定量(天狼星红染色法)、免疫组织化学(α-SMA 染色)和纤维化相关基因的 qRT-PCR/Western blotting 分析 [3] 3. 小鼠癫痫模型:使用雄性 ICR 小鼠(20-25 g);对于 PTZ 诱导的癫痫模型,在腹腔注射 PTZ(80 mg/kg)前 30 分钟,腹腔注射溶于 10% DMSO + 90% 生理盐水的 Y-27632(5、10、20、30 mg/kg),或给予赋形剂;记录癫痫发作潜伏期、严重程度(评分 0-5)和持续时间 30 分钟;对于红藻氨酸模型,在腹腔注射红藻氨酸(30 mg/kg)前 30 分钟腹腔注射 Y-27632(20 mg/kg),并计数 2 小时内的癫痫发作次数 [4] |
| 药代性质 (ADME/PK) |
在大鼠中,Y-27632 的口服生物利用度为 58%,血浆达峰时间 (Tmax) 为 1.2 小时(10 mg/kg 口服),血药浓度峰值 (Cmax) 为 1.2 μg/mL,末端半衰期 (t₁/₂) 为 2.8 小时,分布容积 (Vd) 为 1.5 L/kg [1]。口服后,Y-27632 可迅速吸收,并能穿过血脑屏障(小鼠给药后 1 小时脑/血浆浓度比为 0.25)[4]。代谢:Y-27632 主要在肝脏中通过羟基化(主要代谢物 M1)和 N-去甲基化(次要代谢物 M2)代谢,70% 的原药在 24 小时内经尿液排出(大鼠 10 mg/kg 口服)[1]。
|
| 毒性/毒理 (Toxicokinetics/TK) |
急性毒性:小鼠腹腔注射Y-27632的LD50为210 mg/kg,口服LD50 = 450 mg/kg [1]
亚慢性毒性:大鼠口服Y-27632(30 mg/kg/天)28天,体重、食物消耗量或血清ALT/AST/BUN/肌酐水平无显著变化;肝脏、肾脏、心脏和脑组织的组织病理学分析显示未见异常病变[1] 血浆蛋白结合率:Y-27632在大鼠血浆中的血浆蛋白结合率为42%,在人血浆中的血浆蛋白结合率为38%(超滤法)[1] 细胞毒性:Y-27632在浓度≤20 μM时对正常体细胞(RASMCs、hADSCs、hCSCs)无显著细胞毒性(MTT法检测细胞活力>90%)[1,2,6];在高浓度(>50 μM)时,可诱导胚胎禽角膜上皮细胞的细胞毒性(活力<60%)[8] |
| 参考文献 |
|
| 其他信息 |
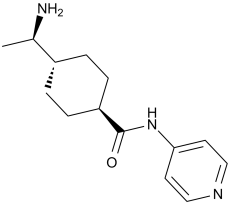
Y-27632 是一种单羧酸酰胺,其结构为反式-[(1R)-1-氨基乙基]环己烷甲酰胺,其中氨基羰基的一个氮原子被吡啶环取代。研究表明,它对 Rho 相关蛋白激酶 (ROCK) 具有抑制活性。它是一种 EC 2.7.11.1(非特异性丝氨酸/苏氨酸蛋白激酶)抑制剂。它是一种单羧酸酰胺,属于吡啶类化合物,也是一种伯氨基化合物。
Y-27632 是 Rho 相关蛋白激酶的抑制剂。它通过抑制钙敏化来影响平滑肌舒张。 背景:p160ROCK 是 Rho 的直接靶点,介导 Rho 诱导的黏着斑和应力纤维的组装。我们之前报道过,Rho 信号通路参与体外肝星状细胞 (HSC) 的活化。本研究旨在验证p160ROCK特异性抑制剂(Y27632)能否预防二甲基亚硝胺(DMN)诱导的大鼠实验性肝纤维化。方法:在首次注射DMN后,每日口服Y27632(30 mg/kg),持续4周。采用图像分析和肝脏胶原蛋白及羟脯氨酸含量测定评估肝纤维化的程度。同时检测肝脏及原代培养的肝星状细胞(HSC)中α-平滑肌肌动蛋白(α-SMA)的表达。采用半定量RT-PCR检测肝脏中I型胶原mRNA的表达。结果:Y27632治疗显著降低了DMN诱导的肝纤维化的发生率,并降低了肝脏胶原蛋白、羟脯氨酸含量以及α-SMA的表达。体外实验也发现 HSC 中 α-SMA 的表达受到抑制。结论:这些发现表明,Rho-ROCK 通路抑制剂可能对肝纤维化具有治疗作用。[3] 在本研究中,我们报道了一个此前未被探索的领域,其中 Y-27632 被鉴定为一种有效的小分子,能够诱导 hIPSC 选择性地向中内胚层谱系分化。Y-27632 通过有效调节细胞骨架和细胞间连接蛋白,诱导 hIPSC 发生类似 EMT 的变化,从而使细胞易于向中内胚层谱系分化。同时,这种对肌动蛋白和 E-钙黏蛋白组织的破坏导致外胚层分化受到抑制。这些结果提出了一种增强人多能干细胞向中内胚层靶细胞类型定向分化的新方法。 [5] 总之,本研究表明,Y-27632预处理可显著降低Caspase-3活性,并保护CSCs免受Dox诱导的细胞凋亡。其潜在机制尚未完全阐明,可能涉及ROCK与Caspase-3、ROCK与LIMK/ADF/Cofilin或ROCK与p-MCL之间的直接和/或间接相互作用(图8)。这些相互作用的总体结果或平衡可能导致对人CSCs的抗凋亡作用。尽管仍有许多问题尚未解答,但Y-27632值得在动物模型中基于干细胞的治疗中进行进一步评估,最终结果可能适用于人体临床试验。[6] 背景:近期使用c-kit+人源心脏干细胞(CSCs)的临床试验在提高心脏功能和改善生活质量方面显示出令人鼓舞的结果。然而,CSCs的效率较低,可能是由于移植后细胞存活率和植入率有限。 Rho 相关蛋白激酶 (ROCK) 抑制剂 Y-27632 显著提高了多种类型细胞(包括干细胞)的存活率、粘附性和迁移能力,这表明 ROCK 介导的细胞凋亡途径具有共同特征,该特征也可能存在于人类癌症干细胞中。本研究旨在验证 Y-27632 预处理人癌症干细胞 (CSC) 是否能保护细胞免受阿霉素 (Dox) 诱导的细胞凋亡。[6] Y-27632 是一种合成吡啶衍生物,也是首个选择性 Rho 相关卷曲螺旋激酶 (ROCK) 小分子抑制剂,由吉富制药(现为安斯泰来制药)于 20 世纪 90 年代开发。[1] 作用机制:Y-27632 与 ROCK 激酶结构域的 ATP 结合口袋结合(I 型竞争性抑制),阻断 ROCK 介导的下游底物(例如 MLC、MYPT1、LIMK)的磷酸化,从而调节肌动蛋白细胞骨架的组织、细胞收缩、迁移和存活。[1] 治疗潜力:Y-27632 Y-27632 在纤维化(肝脏、肾脏)、心血管疾病(血管痉挛、高血压)、神经系统疾病(癫痫、神经退行性疾病)和干细胞生物学(分化、存活)方面具有临床前疗效;它被广泛用作基础研究的工具化合物,但由于在治疗剂量下体内疗效有限,尚未获准用于临床[1,3,4,5]。 化学性质:Y-27632 的分子式为 C₁₄H₂₁N₃O·2HCl,分子量为 336.26 g/mol,logP = 1.2,可溶于水 (100 mM) 和 DMSO (50 mM) [1]。 |
| 分子式 |
C14H21N3O
|
|
|---|---|---|
| 分子量 |
247.34
|
|
| 精确质量 |
247.17
|
|
| 元素分析 |
C, 67.98; H, 8.56; N, 16.99; O, 6.47
|
|
| CAS号 |
146986-50-7
|
|
| 相关CAS号 |
Y-27632 dihydrochloride;129830-38-2;
331752-47-7 (HCl hydrate); 138381-45-0 (racemate HCl); 146986-50-7; 310898-86-3 (recamte free base)
|
|
| PubChem CID |
448042
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.1±0.1 g/cm3
|
|
| 沸点 |
462.6±15.0 °C at 760 mmHg
|
|
| 闪点 |
233.6±20.4 °C
|
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
|
| 折射率 |
1.579
|
|
| LogP |
1.54
|
|
| tPSA |
68.01
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
3
|
|
| 可旋转键数目(RBC) |
3
|
|
| 重原子数目 |
18
|
|
| 分子复杂度/Complexity |
268
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
O=C([C@@H]1CC[C@]([C@H](N)C)([H])CC1)NC2=CC=NC=C2
|
|
| InChi Key |
IYOZTVGMEWJPKR-IJLUTSLNSA-N
|
|
| InChi Code |
InChI=1S/C14H21N3O/c1-10(15)11-2-4-12(5-3-11)14(18)17-13-6-8-16-9-7-13/h6-12H,2-5,15H2,1H3,(H,16,17,18)/t10-,11-,12-/m1/s1
|
|
| 化学名 |
(1R,4r)-4-((R)-1-aminoethyl)-N-(pyridin-4-yl)cyclohexanecarboxamide
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: (1). 本产品在运输和储存过程中需避光。 (2). 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (10.11 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (10.11 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (10.11 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 2 mg/mL (8.09 mM) in Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶 (<60°C). *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.0430 mL | 20.2151 mL | 40.4302 mL | |
| 5 mM | 0.8086 mL | 4.0430 mL | 8.0860 mL | |
| 10 mM | 0.4043 mL | 2.0215 mL | 4.0430 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT05309135 | COMPLETED | Drug:HCEC-1 | Corneal Edema | Aurion Biotech | 2022-03-24 | Phase 1 |
| NCT06041256 | ACTIVE,NOT RECRUITING | Combination Product:AURN001 | Corneal Edema Corneal Endothelial Dysfunction |
Aurion Biotech | 2023-10-18 | Phase 1 Phase 2 |
|
|
|
 HA-100 dihydrochloride
HA-100 dihydrochloride
 ROCK-IN-7
ROCK-IN-7
 ROCK-IN-9
ROCK-IN-9
 ROCK-IN-6
ROCK-IN-6
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA



 463611831
463611831
