| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
Natural product/Steroids; VEGF-VEGF-R2-Akt
(E)-Guggulsterone targets multiple receptors and enzymes. It is an antagonist of the farnesoid X receptor (FXR) with IC50 values of 15 µM (for CDCA-induced FXR activation) and 24.06 µM. It binds to mineralocorticoid receptor (MR) with Ki ≈ 35 nM, which is >100 times more potent than its affinity for FXR. It also binds to androgen receptor (AR), glucocorticoid receptor (GR), and progesterone receptor (PR) with Ki values ranging from 224 to 315 nM. In cell-based assays, (E)-Guggulsterone acts as an antagonist of AR, GR, and MR, but as an agonist of PR, with very low agonist activity on estrogen receptor alpha (ERα, EC50 > 5000 nM). It also inhibits CYP2C9 (19 μM) and CYP2C19 (2.1 μM). |
|---|---|
| 体外研究 (In Vitro) |
GugguLsterone(0.5 -20 μM;24 小时)抑制 TREM-1、TLR4 和 TNF-α 的产生以及 LPS 对 IκBα 和 NF-κB p65 的磷酸化 [2]。
Guggulsterone以剂量和激活依赖的方式抑制LX-2细胞生长。这种生长抑制是由于诱导HSC凋亡,这是通过激活c-Jun N端激酶和线粒体凋亡信号介导的。此外,guggulsterone调节Akt和腺苷一磷酸活化蛋白激酶的磷酸化,随后证明这是guggulseterone诱导的HSC生长抑制的原因。Guggulsterone抑制LX-2细胞中NF-κB的活化,NF-κB是HSC活化的主要介质之一。事实上,guggulsterone降低了这些细胞中胶原蛋白α1的合成和α-平滑肌肌动蛋白的表达。[3] >(E)-Guggulsterone抑制多种肿瘤细胞的生长,并通过下调抗凋亡基因产物(IAP1、xIAP、Bfl-1/A1、Bcl-2、cFLIP和survivin)、调节细胞周期蛋白(cyclin D1和c-Myc)、激活caspases和JNK以及抑制Akt来诱导凋亡。它降低CDCA诱导的FXR激活,E-guggulsterone的IC50为15 μM。在人乳腺上皮MCF10A细胞中,(E)-Guggulsterone(5-25 μM,0-12小时)激活Nrf2信号通路,诱导HO-1表达,并导致适度的细胞内ROS累积。在Huh-7细胞中,它通过Nrf2/HO-1通路活化及抗病毒干扰素应答恢复,表现出强效抗登革热病毒活性(0-20 μM,3天)。它对枯草芽孢杆菌、金黄色葡萄球菌和铜绿假单胞菌具有抗菌作用(3.2 mM,24小时),抑菌圈直径分别为14 mm、14 mm和11 mm。在小鼠及人肝细胞中,(E)-Guggulsterone(1-20 μM)选择性激活ERα(不激活ERα亚型),诱导Cyp3a11与CYP3A4表达。它抑制Cu²⁺介导的LDL脂质过氧化及氧自由基生成(5-20 μM),有效阻断酶促与非酶促脂质过氧化过程。在人肝细胞癌HepG2细胞中,guggulsterone(0、35、50、75 μmol/L,24小时)改变TGF-β1、TNF-α和VEGF水平。Guggulsterone(0.5-20 μM,24小时)抑制LPS诱导的TREM-1、TLR4和TNF-α表达以及IκBα和NF-κB p65的磷酸化。 |
| 体内研究 (In Vivo) |
GugguLsterone(小鼠;100 mg/kg,每天一次,持续 8 天)显着增加野生型小鼠中 TNBS 诱导的膀胱炎的发生率 [2]。
与对照组小鼠或用低剂量guggulsterone 治疗的小鼠相比,高剂量guggulsterone 显著降低了胶原沉积的程度和活化HSC凋亡的百分比。结论:这些结果表明,guggulsterone通过抑制NF-κB活化和诱导凋亡来抑制HSC活化和存活。因此,guggulsterone 可作为慢性肝病的抗纤维化药物。[3] (E)-Guggulsterone(口服,100 mg/kg,每日一次,共8天)显著提高了TNBS诱导结肠炎的野生型小鼠的存活率。它抑制DSS诱导的小鼠结肠炎,表现为临床疾病活动评分、结肠长度和组织学的改善。在大鼠异丙肾上腺素诱导的心肌缺血模型中,guggulsterone(50 mg/kg口服)显著保护心脏损伤,表现为血液和心脏生化参数的逆转。在内毒素诱导的葡萄膜炎大鼠模型中,guggulsterone(30 mg/kg,腹腔注射)阻止了房水中浸润细胞、总蛋白、MMP-2、NO和PGE2的增加,还阻止了眼组织中MMP-2、iNOS、Cox-2、IκB和NF-κB的表达。通过抑制CDCA诱导的FXR反式激活,(E)-Guggulsterone降低高胆固醇饮食啮齿动物的低密度脂蛋白胆固醇和甘油三酯水平。 |
| 酶活实验 |
在炎症性肠病(IBD)患者的结肠中,表达髓样细胞1(TREM-1)的肠巨噬细胞上表达的触发受体显著增加。我们在这里重点研究了guggulsterone作为人类IBD潜在治疗分子对结肠炎巨噬细胞调节的影响,并探讨了其潜在机制。检测巨噬细胞中的基因表达,并使用HT-29细胞进行伤口愈合试验。通过向结肠中施用2,4,6-三硝基苯磺酸(TNBS)诱导野生型和IL-10-、Toll样受体4(TLR4)和髓系分化初级反应88(MyD88)缺陷小鼠的结肠炎。在体外和体内实验中,guggulsterone抑制了TREM-1刺激放大的肠道炎症,其中涉及抑制NF-κB、激活蛋白-1和蛋白酶体途径。在TNBS诱导的结肠炎模型中,guggulsterone降低了疾病活动指数评分和TREM-1表达,刺激了IL-10的产生,并提高了野生型小鼠的存活率。在IL-10-、TLR4-和MyD88缺陷小鼠中没有观察到这些作用。Guggulsterone还抑制了M1极化,但在IBD患者和小鼠的巨噬细胞中诱导了M2表型。这些发现表明,guggulsterone通过TREM-1抑制阻断巨噬细胞的过度活化,并通过TLR4信号通路介导的IL-10诱导M2极化。此外,本研究为guggulsterone治疗IBD的治疗潜力提供了新的理论基础。新的和值得注意的是,我们发现guggulsterone减弱了髓系细胞1(TREM-1)上表达的触发受体介导的巨噬细胞过度活化,并使巨噬细胞向M2表型极化。这是由IL-10和部分Toll样受体4信号通路介导的。总体而言,这些数据支持guggulsterone作为一种天然植物甾醇调节结肠炎中的巨噬细胞表型,这可能在炎症性肠病治疗中具有新的治疗重要性[2]。
描述了以下实验方案:FXR拮抗实验 – 测量鹅去氧胆酸诱导的FXR激活,(E)-Guggulsterone降低CDCA诱导的FXR激活,IC50为15 µM。甾体受体结合实验 – (E)-和(Z)-guggulsterone均结合盐皮质激素受体,Ki ≈ 35 nM,结合雄激素受体、糖皮质激素受体和孕激素受体的Ki值为224至315 nM。在基于细胞的功能共转染实验中,guggulsterone作为AR、GR和MR的拮抗剂,但作为PR的激动剂。CYP酶抑制实验 – (E)-Guggulsterone抑制CYP2C9的IC50为19 μM,抑制CYP2C19的IC50为2.1 μM。 |
| 细胞实验 |
描述了以下基于细胞的实验方案:MCF10A细胞Nrf2激活实验 – 人乳腺上皮MCF10A细胞用(E)-Guggulsterone(5-25 μM,0-12小时)处理,评估Nrf2信号通路激活和HO-1表达。Huh-7细胞抗登革热病毒实验 – Huh-7细胞用(E)-Guggulsterone(0-20 μM,3天)处理,通过Nrf2/HO-1通路活化评估抗DENV活性。抗菌纸片扩散实验 – (E)-Guggulsterone(3.2 mM,24小时)对枯草芽孢杆菌、金黄色葡萄球菌和铜绿假单胞菌进行测试,测量抑菌圈直径。肝细胞CYP3A诱导实验 – 小鼠和人肝细胞用(E)-Guggulsterone(1-20 μM)处理,评估ERα激活和Cyp3a11/CYP3A4表达。LDL脂质过氧化抑制实验 – (E)-Guggulsterone(5-20 μM)测试对Cu²⁺介导的LDL脂质过氧化及氧自由基生成的抑制作用。HepG2细胞凋亡实验 – 人肝细胞癌HepG2细胞用guggulsterone(0、35、50、75 μmol/L,24小时)处理,通过ELISA测量TGF-β1、TNF-α和VEGF水平。LPS诱导炎症实验 – 细胞用guggulsterone(0.5-20 μM,24小时)处理,评估对TREM-1、TLR4和TNF-α表达以及IκBα和NF-κB p65磷酸化的抑制。
|
| 动物实验 |
本研究使用了35只6周龄的雄性白鼠,体重在20-30克之间。所有小鼠均饲养于通风良好的塑料笼中,饲养环境温度、湿度和光照/黑暗周期均相对可控。小鼠可自由摄取购自当地市场的标准商业饲料和自来水。小鼠随机分为5组,每组7只,具体如下:第一组:饲喂标准商业饲料,作为对照组。第二组:饲喂特制高脂饲料(HFD)12周,以诱导非酒精性脂肪肝。第三组:饲喂添加500 ppm古古甾酮的高脂饲料12周。第四组:本组小鼠饲喂含1000 ppm古古甾酮的高脂饲料,持续12周。第五组:本组小鼠饲喂含2000 ppm古古甾酮的高脂饲料,持续12周。所有组别的小鼠每周常规测量一次体重,并在实验开始时(即实验开始前)再次测量体重。实验结束时,各组动物禁食过夜后,用乙醚麻醉,通过心脏穿刺采集心脏血,然后处死动物,取出肝脏并称重,计算肝脏指数(肝脏重量以毫克计,除以最后一次体重)。[4] 古古甾酮是古古脂的活性成分,古古脂是一种源自没药树(Commiphora mukul)的阿育吠陀药物,据报道具有降血脂活性。本研究在大鼠中研究了Z-古古甾酮(1a)及其代谢物E-古古甾酮(1b)的药代动力学,分别采用口服(50 mg kg−1)和静脉注射(18 mg kg−1)的方式给药。观察到在给药后的大鼠血清样本中,1a异构化为1b。[3] 静脉注射后,血清中古古甾酮的浓度呈现双指数消除曲线。异构体 1a 和 1b 的平均末端半衰期分别为 10.02 ± 4.74 小时和 9.24 ± 3.32 小时。1a 和 1b 的系统清除率和 AUC 值分别为 0.71 Lh⁻¹、4.9 μg·h·mL⁻¹ 和 1.04 Lh⁻¹、3–65 μg·h·mL⁻¹。口服给药后,浓度-时间曲线呈单指数衰减,1a 和 1b 的 Cmax、末端半衰期、清除率和 AUC 值分别为 1.07 μg·mL⁻¹、4.48 小时、1.76 Lh⁻¹、5.95 μg·mL⁻¹ 和 0.97 μg·mL⁻¹、3.56 小时、2.24 Lh⁻¹ 和 4.75 μg·h·mL⁻¹。绝对值口服给药后,母体化合物(1a)的生物利用度为42.9%。[3]
描述了以下动物实验方案:结肠炎模型(TNBS诱导) – TNBS诱导结肠炎的野生型小鼠口服(E)-Guggulsterone(100 mg/kg,每日一次,共8天),测量存活率。结肠炎模型(DSS诱导) – DSS诱导的小鼠结肠炎,通过临床疾病活动评分、结肠长度和组织学评估guggulsterone的治疗效果。心肌缺血模型 – 异丙肾上腺素诱导的大鼠心肌缺血;(E)-Guggulsterone(E和Z异构体)以50 mg/kg口服给药,通过血液和心脏生化参数的逆转评估心脏损伤保护作用。内毒素诱导葡萄膜炎模型 – 通过皮下注射脂多糖(150 μg)至Lewis大鼠诱导EIU;guggulsterone以30 mg/kg体重腹腔注射。24小时后摘除眼球,收集房水,测量浸润细胞、总蛋白、MMP-2、NO、PGE2和炎症细胞因子表达。高脂血症模型 – 高胆固醇饮食喂养的啮齿动物接受(E)-Guggulsterone处理,测量LDL胆固醇和甘油三酯水平以评估降血脂效果。 |
| 药代性质 (ADME/PK) |
(E)-Guggulsterone口服有效。两种异构体与大鼠血浆蛋白高度结合(>95%结合率)。口服给药后,血浆浓度迅速下降,E-异构体的终末半衰期为0.63 ± 0.25小时。E-异构体的清除率为2.79 ± 0.73 L/h/kg。在大鼠肝微粒体中,E-异构体的体外固有清除率为33.34 ± 0.51 μL/min/mg蛋白。首过代谢似乎是导致guggulsterone在大鼠中生物利用度低的原因。(E)-Guggulsterone在人、猴、兔及大鼠血浆中呈高度蛋白结合(>96%),并能促进肝微粒体氧化代谢。ADME预测显示无Lipinski和Veber规则违反。该化合物可能抑制CYP2C19和CYP2C9,但不能抑制CYP1A2、CYP2D6和CYP3A4。
|
| 毒性/毒理 (Toxicokinetics/TK) |
古古鲁脂有几个报告的副作用,包括广泛性麻疹样红斑性丘疹、局限于手臂的斑疹、面部肿胀和红斑伴烧灼感、瘙痒以及小腿大疱性病变,伴有头痛、肌痛和瘙痒。计算机模拟预测显示guggulsterone无肝毒性、细胞毒性或致突变性效应。(E)-Guggulsterone诱导人CYP3A的表达。大剂量时可能导致健康损害,并对眼睛、皮肤或呼吸器官造成刺激。
|
| 参考文献 |
[1]. Guggulsterone for Chemoprevention of Cancer. Curr Pharm Des. 2016;22(3):294-306.
[2]. Protective effects of guggulsterone against colitis are associated with the suppression of TREM-1 and modulation of macrophages. Am J Physiol Gastrointest Liver Physiol. 2018 Jul 1;315(1):G128-G139. [3]. Guggulsterone attenuates activation and survival of hepatic stellate cell by inhibiting nuclear factor kappa B activation and inducing apoptosis. J Gastroenterol Hepatol. 2013 Dec;28(12):1859-68. [4]. Possible Amelioration of the Severity of Nutritional Steatohepatitis by Guggulsterone in Mice. Iraqi J Pharm Sci, Vol.28(1) 2019. DOI: https://doi.org/10.31351/vol28iss1pp17-23; https://bijps.uobaghdad.edu.iq/index.php/bijps/article/view/801; https://pdfs.semanticscholar.org/fed5/b2b899b009d9a009406450b1d111bb67f233.pdf |
| 其他信息 |
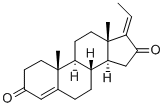
E-古古甾酮是一种3-羟基类固醇,具有雄激素活性。
已有报道称,没药树(Commiphora mukul)和没药树(Commiphora wightii)中含有E-古古甾酮,并有相关数据可供参考。 (E)-Guggulsterone是一种天然存在的植物甾醇(3-羟基甾体),存在于Commiphora mukul和Commiphora wightii中。也称为(-)-(E)-Guggulsterone,分子式为C21H28O2,分子量为312.45。它能有效逆转多种人类癌细胞系的多药耐药性,延长化疗的疗效。Guggulsterone是FXR的竞争性拮抗剂,在体外和体内均有效。它通过Nrf2激活诱导HO-1表达,上调抗病毒干扰素应答,从而抑制登革热病毒复制。该化合物正在被研究用于癌症的化学预防,并在头颈部鳞状细胞癌细胞系中显示出活性,EC50值范围为5至8 μM,可诱导凋亡和细胞周期阻滞,抑制侵袭,并增强厄洛替尼、西妥昔单抗和顺铂的疗效。合成guggulsterone的心脏保护和抗氧化活性与天然guggulsterone相当。Guggulsterone及其两种异构体在5-20 mM浓度下抑制金属离子诱导的人低密度脂蛋白和大鼠肝微粒体的脂质氧化降解。需要进一步研究以评估guggulsterone的潜在生殖毒性。 |
| 分子式 |
C21H28O2
|
|---|---|
| 分子量 |
312.44582
|
| 精确质量 |
312.208
|
| 元素分析 |
C, 80.73; H, 9.03; O, 10.24
|
| CAS号 |
39025-24-6
|
| 相关CAS号 |
39025-23-5 (Z-Guggulsterone); 95975-55-6; 39025-24-6 (E-Guggulsterone)
|
| PubChem CID |
6439929
|
| 外观&性状 |
Typically exists as White to off-white solids at room temperature
|
| 密度 |
1.1±0.1 g/cm3
|
| 沸点 |
463.3±45.0 °C at 760 mmHg
|
| 熔点 |
170-171.5°
|
| 闪点 |
172.3±25.7 °C
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
| 折射率 |
1.557
|
| LogP |
3.65
|
| tPSA |
34.14
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
2
|
| 可旋转键数目(RBC) |
0
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
640
|
| 定义原子立体中心数目 |
5
|
| SMILES |
C/C=C1/C2(C)C(C3C(CC2)C2(C)C(=CC(=O)CC2)CC3)CC/1=O
|
| InChi Key |
WDXRGPWQVHZTQJ-AUKWTSKRSA-N
|
| InChi Code |
InChI=1S/C21H28O2/c1-4-16-19(23)12-18-15-6-5-13-11-14(22)7-9-20(13,2)17(15)8-10-21(16,18)3/h4,11,15,17-18H,5-10,12H2,1-3H3/b16-4-/t15-,17+,18+,20+,21-/m1/s1
|
| 化学名 |
(8R,9S,10R,13S,14S,17E)-17-ethylidene-10,13-dimethyl-1,2,6,7,8,9,11,12,14,15-decahydrocyclopenta[a]phenanthrene-3,16-dione
|
| 别名 |
(E)-Guggulsterone; E-Guggulsterone; 39025-24-6; Guggulsterone E; Guggulsterone; trans-Guggulsterone; Guggulsterones E; Guggulsterone, (E)-;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~25 mg/mL (~80.01 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2 mg/mL (6.40 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.0 mg/mL 澄清 DMSO 储备液加入900 μL 玉米油中,混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2005 mL | 16.0026 mL | 32.0051 mL | |
| 5 mM | 0.6401 mL | 3.2005 mL | 6.4010 mL | |
| 10 mM | 0.3201 mL | 1.6003 mL | 3.2005 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT01492998 | Terminated | Other: guggulsterone, a natural FXR antagonist. | Chronic Hepatitis C | Hospices Civils de Lyon | 2010-01 | Not Applicable |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
