| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
VEGF-R2
Farnesoid X receptor (FXR) antagonist[2] (Z)-Guggulsterone (CAS: 39025-23-5) is a broad-spectrum steroid receptor ligand that functions primarily as an antagonist of the farnesoid X receptor (FXR). Regarding steroid hormone receptors, it acts as an antagonist of the mineralocorticoid receptor (MR, Ki = 37 nM), progesterone receptor (PR, Ki = 224 nM), and glucocorticoid receptor (GR, Ki = 252 nM), while serving as a weak agonist of the androgen receptor (AR, Ki = 315 nM). Furthermore, this compound inhibits angiogenesis by suppressing the VEGF-VEGFR2-Akt signaling axis. |
|---|---|
| 体外研究 (In Vitro) |
在 HUVEC 中,(Z)-GugguLsterone(10、20 μM;24 或 48 小时)可降低 VEGF-R2 蛋白水平 [1]。通过 FXR 介导的 ACE2 调节,(Z)-Guggulsterone (10 μM;24) 可减少初级气道,扰乱类器官中的 ACE2 和 SHP 水平,并减少许多细胞类型中的 SARS-CoV-2 感染 [2]。
z-guggulsterone治疗以浓度和时间依赖的方式抑制人脐静脉内皮细胞(HUVEC)的毛细血管样管形成(体外新生血管形成)以及HUVEC和DU145人前列腺癌症细胞的迁移。guggulsterone的z和E异构体作为HUVEC管形成的抑制剂似乎是等效的[1]。 前期研究发现,z-guggulsterone是印度阿育吠陀药用植物commiora mukul的一种成分,通过引起细胞凋亡抑制人前列腺癌细胞的生长。我们现在报道了一种新的对z-guggulsterone的反应,涉及体外和体内血管生成的抑制。z-谷固酮治疗抑制人脐静脉内皮细胞(HUVEC)的毛细血管样管形成(体外新生血管),并以浓度和时间依赖性方式抑制HUVEC和DU145人前列腺癌细胞的迁移。谷谷固酮的z-和e -异构体似乎具有抑制HUVEC管形成的同等效力。z-guggulsterone介导的体外血管生成抑制与抑制促血管生成生长因子(如血管内皮生长因子(VEGF)和粒细胞集落刺激因子)的分泌、下调VEGF受体2 (VEGF- r2)蛋白水平和Akt失活相关。z-guggulsterone介导的对DU145细胞迁移的抑制通过敲低VEGF-R2蛋白水平而增强。在DU145细胞中,组成活性Akt的异位表达对z-guggulsterone介导的细胞迁移抑制[1]具有保护作用。 用10 µM的FXR拮抗剂(Z)-Guggulsterone处理,可降低FXR信号传导,减少FXR在ACE2启动子上的结合,并在原代人胆管细胞、气道和肠道类器官中在转录和蛋白水平上下调ACE2的表达。这种ACE2表达的下调 subsequently 降低了这些类器官模型对SARS-CoV-2感染的易感性。[2] 在用于模拟基线FXR激活的、经生理水平胆汁酸CDCA(一种FXR激动剂)处理的胆囊胆管细胞类器官中,与10 µM (Z)-Guggulsterone共处理可减少SARS-CoV-2感染,通过感染后24小时的病毒RNA定量和病毒刺突蛋白免疫荧光检测证实。[2] 在胆管细胞类器官中使用shRNA敲低FXR后,CDCA处理无法再上调ACE2,并且病毒感染被抑制,且该抑制效果不依赖于CDCA、UDCA或ZGG的处理。在FXR敲低后,用(Z)-Guggulsterone处理对病毒感染没有额外影响,证实其抗病毒作用是通过FXR介导的。[2] 在通过基因工程稳定过表达ACE2(且不受FXR调控)的HEK293T细胞中,用(Z)-Guggulsterone处理不影响SARS-CoV-2复制,证实其抗病毒作用 specifically 依赖于其通过FXR调节ACE2表达的能力。[2] 含有ACE2启动子中FXR反应元件(IR-1)的荧光素酶报告基因实验显示,用50 µM (Z)-Guggulsterone处理会降低与该元件相关的转录活性,而对IR-1位点进行定点突变则消除了此效应,证实了FXR结合的特异性。[2] |
| 体内研究 (In Vivo) |
(Z)-Guggulsterone(二氧化硅;1 mg;每周 5 次)可显着降低湿重和肿瘤体积 [1]。
在DU145 Matrigel塞试验中,对雄性裸鼠口服1mg z-guggulsterone/d(五次/周)抑制了体内血管生成,其证据是肿瘤负荷、微血管面积(血管生成标志物因子VIII和CD31染色)和VEGF-R2蛋白表达在统计学上显著降低。总之,本研究表明,z-guggulsterone通过抑制VEGF-VEGF-R2-Akt信号轴来抑制血管生成。总之,我们的研究结果为z-guggulsterone对前列腺癌症的疗效的进一步临床前和临床研究提供了令人信服的理由[1]。 给雄性裸鼠口服(Z)-Guggulsterone(每只小鼠1 mg,约40 mg/kg,每周五次),从含有DU145细胞的Matrigel栓皮下植入前2周开始,持续到植入后2周,显著抑制了体内血管生成。与溶剂对照组相比,肿瘤体积和湿重显著减少证明了这一点。[1] 对切除的Matrigel栓进行免疫组化分析表明,与对照组相比,给予(Z)-Guggulsterone显著降低了微血管面积(基于血管生成标记物因子VIII和CD31的染色)并降低了VEGF-R2蛋白表达。[1] |
| 酶活实验 |
z-guggulsterone介导的体外血管生成抑制与抑制促血管生成生长因子[如血管内皮生长因子(VEGF)和粒细胞集落刺激因子]的分泌、下调VEGF受体2(VEGF-R2)蛋白水平和Akt失活相关。z-guggulsterone介导的对DU145细胞迁移的抑制通过敲低VEGF-R2蛋白水平而增加。DU145细胞中组成型活性Akt的异位表达对z-guggulsterone介导的细胞迁移抑制具有保护作用[3]。
对胆管细胞类器官进行了染色质免疫沉淀随后定量PCR分析。在收集细胞前,用含有100 µM CDCA、UDCA或(Z)-Guggulsterone的新鲜培养基孵育细胞2小时。裂解液与FXR抗体或非免疫IgG一起孵育过夜。纯化免疫沉淀的DNA,并使用针对ACE2启动子上FXR结合位点设计的引物进行qPCR分析。FXR激动剂CDCA促进了FXR与ACE2启动子的结合,而FXR抑制剂UDCA和(Z)-Guggulsterone则减少了这种结合。[2] 采用荧光素酶报告基因实验评估FXR转录活性。将含有ACE2或SHP基因中FXR IR-1反应元件的片段克隆到pGL3-启动子荧光素酶载体中。通过定点突变生成IR-1位点的突变体。将这些报告基因载体与FXR表达质粒共转染到HEK293细胞中。转染后24小时,用含有50 µM CDCA、UDCA或(Z)-Guggulsterone的新鲜培养基处理细胞8小时。测量荧光素酶活性并归一化至空pGL3载体。实验表明,CDCA通过ACE2 IR-1元件增加了转录活性,而UDCA和(Z)-Guggulsterone降低了此活性。IR-1位点的突变则消除了该活性。[2] |
| 细胞实验 |
蛋白质印迹分析 [1]
细胞类型:血管内皮生长因子 (VEGF) 测试浓度: 10、20 μM 孵育持续时间:24 或 48 小时 实验结果:导致 HUVEC 中 VEGF-R2 蛋白水平下降。 细胞培养与细胞活力检测 [1] 人脐静脉内皮细胞(HUVEC)购自Clonetics公司,培养于添加5%胎牛血清的内皮细胞生长培养基-2(EGM2 MV SingleQuots)中。DU145细胞单层培养方法如我们先前所述。香胶甾酮各异构体的母液用DMSO配制,并用完全培养基稀释。对照组加入等体积DMSO(终浓度<0.2%)。Z型和E型香胶甾酮对HUVEC活力的影响通过磺酰罗丹明B法测定,方法如我们先前所述。 体外毛细血管样管腔形成与迁移实验 [1] Z型和E型香胶甾酮对体外血管生成的影响通过管腔形成实验评估,方法如我们先前报道。接种于Matrigel的HUVEC可分化形成毛细血管样管状结构。部分管腔形成实验中,HUVEC在存在或不存在1 μmol/L Akt-1/2抑制剂条件下,暴露于20 μmol/L Z型香胶甾酮处理24小时。采用Transwell Boyden小室(聚碳酸酯滤膜孔径8 μm)评估Z型香胶甾酮对HUVEC或DU145细胞体外迁移的影响,方法如我们先前所述。部分迁移实验中,HUVEC或DU145细胞在存在或不存在1 μmol/L Akt-1/2抑制剂条件下,用20 μmol/L Z型香胶甾酮处理24小时。 免疫印迹分析 [1] 总Akt、磷酸化Akt及VEGF-R2的免疫印迹检测方法如我们先前所述。简言之,用指定浓度Z型香胶甾酮处理HUVEC或DU145细胞特定时间后,收集悬浮与贴壁细胞。细胞裂解液制备方法如前述。裂解蛋白经6%-10% SDS-PAGE分离后转印至聚偏二氟乙烯膜。经一抗和二抗处理后,采用增强化学发光法显色免疫反应条带。剥离印迹膜后用抗肌动蛋白抗体复染以校正上样量差异。通过条带光密度扫描定量蛋白水平变化。每种蛋白的免疫印迹实验均使用独立制备的裂解液重复至少两次。 生长因子、白细胞介素及基质金属蛋白酶分析 [1] 将HUVEC或DU145细胞(2×10⁵个)接种于24孔板,孵育过夜使贴壁。细胞用指定浓度Z型香胶甾酮或DMSO(对照)处理24及48小时后,收集培养上清,采用商品化ELISA试剂盒检测VEGF、EGF、G-CSF、FGF、IL-12、IL-17、MMP-2和MMP-9的分泌水平,方法如我们先前所述。 VEGF-R2的RNA干扰 [1] 采用靶向VEGF-R2的小干扰RNA(siRNA)进行基因沉默。非特异性对照siRNA购自Qiagen公司。转染时,将DU145细胞(5×10⁴个)接种于6孔板过夜贴壁,按制造商推荐方案使用OligofectAMINE转染200 nmol/L对照siRNA或VEGF-R2靶向siRNA。转染24小时后,用DMSO(对照)或20 μmol/L Z型香胶甾酮处理细胞24小时,收集细胞进行迁移实验和免疫印迹分析,方法同前。 组成型活性Akt的异位表达 [1] 采用Fugene 6转染试剂将编码组成型活性Akt-1(Myr-Akt1-HA)的pCMV6载体或空载体瞬时转染DU145细胞。简言之,将DU145细胞以2×10⁵个/mL密度接种于6孔板过夜贴壁,转染组成型活性Akt表达载体或空载体。转染24小时后,用20 μmol/L Z型香胶甾酮或DMSO(对照)处理细胞24小时,检测总Akt/磷酸化Akt水平并进行迁移实验。 染色质免疫沉淀(ChIP)实验 [2] 每组ChIP实验使用约6×10⁶个细胞。细胞在收集前2小时用含100 μM CDCA(鹅去氧胆酸)、UDCA(熊去氧胆酸)或Z型香胶甾酮(ZGG)的新鲜培养基孵育。使用True Micro ChIP试剂盒按说明书操作:1)预清除后,裂解液与FXR抗体(附表1)或非免疫IgG 4℃孵育过夜;2)通过MicroChip DiaPure柱纯化免疫沉淀DNA;3)采用ΔΔCt法进行qPCR分析(引物序列见附表3)。阳性对照使用FXR靶基因OSTα(即SLC51A)FXRE侧翼引物,阴性对照使用ACE2启动子非FXRE区域引物。结果以非免疫IgG ChIP对照的富集程度进行标准化。 荧光素酶报告基因实验 [2] 以人类基因组DNA为模板,扩增ACE2基因和SHP基因(即NR0B2)中FXRE IR-1片段,克隆至pGL3-promoter荧光素酶载体。通过定点突变构建ACE2和SHP IR-1突变体(引物序列见附表4)。使用TransIT-293转染试剂将报告基因载体与FXR表达质粒共转染HEK293细胞。转染24小时后,用50 μM CDCA、UDCA或ZGG处理8小时。采用GLO-Luciferase Reporter系统检测荧光素酶活性,结果以空pGL3载体标准化。 细胞毒性与活力检测 [2] 类器官分别用0.1-100 μM CDCA、UDCA或ZGG处理:1)台盼蓝染色后用Countess II细胞计数仪计数活细胞百分比;2)10 μM处理组采用PrestoBlue刃天青法,使用SpectraMax M2酶标仪(SoftMax Pro 5.4.4软件)检测细胞活力。 SARS-CoV-2复制荧光素酶报告系统 [2] 构建方法如文献28所述:1)HEK293T报告细胞稳定表达ACE2、海肾荧光素酶(Rluc)和SARS-CoV-2木瓜样蛋白酶激活的环状排列萤火虫荧光素酶(FFluc);2)接种于96孔板后,用不同浓度CDCA、UDCA或ZGG处理,以MOI=0.01感染SARS-CoV-2;3)阳性对照为瑞德西韦(RdRp抑制剂)和REGN-COV2(中和抗体组合);4)24小时后裂解细胞,采用Dual-Glo试剂盒测定FFluc/Rluc活性比,通过GraphPad Prism软件进行4参数logistic曲线拟合分析。 使用了原代人胆囊胆管细胞类器官、气道类器官和肠道类器官。为了调节FXR活性,将类器官与终浓度为10 µM的CDCA,或10 µM CDCA与10 µM UDCA或(Z)-Guggulsterone的组合一起孵育。[2] 对于ACE2表达分析,按上述方法处理类器官。提取RNA,并通过定量PCR测量ACE2 mRNA水平,使用HMBS或GAPDH作为持家基因进行归一化。使用特异性ACE2抗体通过免疫荧光评估蛋白表达。[2] 对于SARS-CoV-2感染实验,类器官先用生理水平的CDCA预处理以模拟基线FXR激活,并在存在或不存在FXR抑制剂的情况下进行。然后以感染复数(MOI)为1的SARS-CoV-2感染类器官2小时。洗涤后,培养感染的类器官并在感染后24小时收集。通过针对SARS-CoV-2 RNA(靶向RdRp基因)的qPCR(归一化至GAPDH)和针对SARS-CoV-2刺突蛋白的免疫荧光染色来定量病毒感染。[2] 对用一定浓度范围的CDCA、UDCA或(Z)-Guggulsterone(0.1 µM – 100 µM)处理的原代类器官进行了细胞毒性和活力测定。使用台盼蓝拒染计数法和基于刃天青的代谢活性测定法评估细胞活力。在实验所用浓度(例如10 µM)下未观察到细胞毒性作用。[2] 使用携带针对人FXR的shRNA的慢病毒颗粒在胆管细胞类器官中进行FXR敲低。对照组类器官接受携带乱序shRNA的慢病毒颗粒。成功转导的类器官用嘌呤霉素进行筛选。在敲低10天后进行评估ACE2表达和SARS-CoV-2感染的实验。[2] |
| 动物实验 |
动物/疾病模型:将含有DU145细胞的Matrigel胶塞皮下植入5-6周龄的雄性裸鼠体内。
剂量:1 mg。 给药途径:口服(po,即灌胃),每周5次。 实验结果:肿瘤体积和湿重均显著降低。 体内Matrigel胶塞试验[1] 采用DU145-Matrigel胶塞试验测定z-古古甾酮对体内血管生成的影响。从Taconic公司购买5-6周龄的雄性裸鼠,随机分为两组,每组5只。在Matrigel胶塞植入前两周,小鼠分别经口灌胃给予0.1 mL载体(PBS)或1 mg/只小鼠溶于0.1 mL PBS中的z-古古甾酮(相当于约40 mg/kg体重),每周五次。将含有3 × 10⁶个DU145细胞的Matrigel胶塞皮下植入每只小鼠的侧腹部。z-古古甾酮和载体的给药持续两周。肿瘤体积采用游标卡尺测量,方法如我们之前所述。每周记录载体对照组和z-古古甾酮治疗组小鼠的体重。同时监测各组小鼠的其他副作用症状,包括食欲和饮水减少以及姿势或运动障碍。Matrigel胶塞植入后14天处死动物。实验结束后,取出对照组和z-古古甾酮处理组小鼠的Matrigel胶塞,并用10%中性缓冲福尔马林固定。将固定后的Matrigel胶塞脱水、石蜡包埋,并切片至4 μm厚。取对照组和z-古古甾酮处理组小鼠的切片进行CD31、VIII因子和VEGF-R2的免疫组织化学分析。使用图像分析软件,基于CD31和VIII因子的免疫染色,对微血管面积进行定量图像分析。 体内Matrigel胶塞血管生成实验:将5-6周龄的雄性裸鼠随机分为对照组和处理组。治疗组小鼠每周五次灌胃给予(Z)-古古甾酮,剂量为每只小鼠1 mg(约40 mg/kg体重),溶于0.1 mL磷酸盐缓冲液(PBS)中。对照组小鼠灌胃给予0.1 mL PBS溶剂。此预处理方案持续两周。随后,将含有3 × 10^6个DU145细胞的Matrigel胶塞皮下植入每只小鼠的侧腹部。植入后继续灌胃给予(Z)-古古甾酮或溶剂两周。定期使用游标卡尺测量肿瘤体积。植入胶塞14天后,处死小鼠。取出Matrigel胶塞,用福尔马林固定,进行石蜡包埋,并切片进行免疫组织化学分析。[1] |
| 药代性质 (ADME/PK) |
一篇引用的大鼠药代动力学研究(论文中的参考文献41)报告称,单次口服50 mg/kg体重的古古甾酮后,血浆中古古甾酮的最大浓度约为3.3 µM。[1]
同一项引用的大鼠研究报告称,(Z)-古古甾酮的口服生物利用度约为43%。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
在体内研究中,对雄性裸鼠口服给予(Z)-古古甾酮(1 mg/只,约40 mg/kg,每周5次,持续4周),与载体对照组相比,未引起体重显著变化。接受治疗的小鼠外观健康,未出现任何不适、活动受限、消化不良或局部反应(如发红或肿胀)的迹象。[1]
体外研究表明,浓度高达20 µM的(Z)-古古甾酮在24小时内未对人脐静脉内皮细胞(HUVEC)的活力产生显著影响。[1] |
| 参考文献 | |
| 其他信息 |
古古甾酮是一种3-羟基类固醇,具有雄激素活性。
已有报道称古古甾酮存在于没药树(Commiphora mukul)和白花没药树(Commiphora wightii)中,并有相关数据报道。 我们之前的研究表明,印度阿育吠陀药用植物没药树(Commiphora mukul)的成分之一——Z-古古甾酮,可通过诱导细胞凋亡抑制人前列腺癌细胞的生长。我们现在报道了Z-古古甾酮的一项新作用,即在体外和体内抑制血管生成。Z-古古甾酮处理以浓度和时间依赖的方式抑制人脐静脉内皮细胞(HUVEC)的毛细血管样管状结构形成(体外新生血管形成)以及HUVEC和DU145人前列腺癌细胞的迁移。古古甾酮的Z-和E-异构体作为HUVEC管状结构形成的抑制剂,其效力似乎相当。体外实验表明,Z-古古甾酮介导的血管生成抑制与促血管生成生长因子(例如血管内皮生长因子 (VEGF) 和粒细胞集落刺激因子)分泌的抑制、VEGF 受体 2 (VEGF-R2) 蛋白水平的下调以及 Akt 的失活相关。VEGF-R2 蛋白水平的敲低增强了 Z-古古甾酮介导的 DU145 细胞迁移抑制作用。在 DU145 细胞中异位表达组成型活性 Akt 可保护细胞免受 Z-古古甾酮介导的细胞迁移抑制作用。向雄性裸鼠灌胃给予1 mg/d(每周五次)z-古古甾酮,可抑制DU145-Matrigel胶塞试验中的体内血管生成,表现为肿瘤负荷、微血管面积(血管生成标志物VIII因子和CD31染色)以及VEGF-R2蛋白表达的统计学显著降低。总之,本研究表明z-古古甾酮通过抑制VEGF-VEGF-R2-Akt信号通路来抑制血管生成。我们的研究结果为进一步开展z-古古甾酮治疗前列腺癌的临床前和临床研究提供了充分的理论依据。[1] 通过调节病毒宿主受体(例如血管紧张素转化酶2 (ACE2))来预防SARS-CoV-2感染,可能代表一种新的COVID-19化学预防方法,可作为疫苗接种的补充[2,3]。然而,ACE2表达的调控机制尚不明确。本研究表明,法尼醇X受体(FXR)是多种受COVID-19影响的组织(包括胃肠道和呼吸系统)中ACE2转录的直接调控因子。我们随后使用非处方药Z-古古甾酮和已过专利期的熊去氧胆酸(UDCA)来降低FXR信号传导,从而下调人肺、胆管细胞和肠道类器官以及小鼠和仓鼠相应组织中的ACE2表达。结果表明,UDCA介导的ACE2下调可降低体外、体内以及离体灌注的人肺和肝脏对SARS-CoV-2感染的易感性。此外,我们还发现UDCA可降低人鼻上皮细胞中ACE2的表达。最后,我们利用回顾性登记数据,发现 UDCA 治疗与 SARS-CoV-2 感染后的积极临床结局之间存在相关性,并在一个独立的肝移植受者验证队列中证实了这些发现。总之,我们证明 FXR 在控制 ACE2 表达中发挥作用,并提供了证据表明,调节该通路可能有助于降低 SARS-CoV-2 感染,为未来的临床试验铺平了道路。[2] 肿瘤血管生成(新生血管形成)是一个高度复杂的过程,受多种促血管生成生长因子及其相应受体的调控。基于本研究的结果,可以合理地得出结论:抑制 VEGF-VEGF-R2-Akt 信号通路可能是 Z-古古甾酮发挥抗血管生成作用的重要机制。以下观察结果支持该结论:(a) z-古古甾酮介导的管状结构形成和迁移抑制与 VEGF 分泌的抑制相关,VEGF 通过受体酪氨酸激酶 VEGF-R2 向正常和肿瘤来源的内皮细胞提供促生存信号;(b) z-古古甾酮治疗下调 VEGF-R2 的蛋白水平;(c) VEGF-R2 蛋白水平的敲低增强了 z-古古甾酮介导的 DU145 细胞迁移抑制;(d) z-古古甾酮抑制 HUVEC 和 DU145 细胞中的 Akt,并且该药物对 HUVEC 管状结构的抑制作用因 Akt 的药理学抑制而增强。然而,Z-古古甾酮降低VEGF分泌或下调VEGF-R2蛋白水平的具体机制尚不明确,需要进一步研究。总之,本研究表明Z-古古甾酮在体外和体内均能抑制血管生成。Z-古古甾酮介导的血管生成抑制与Akt失活、生长因子(VEGF和G-CSF)、IL-17和MMP-2分泌抑制以及VEGF-R2蛋白表达下调有关。[1] (Z)-古古甾酮是一种非处方植物甾醇,也是一种FXR拮抗剂。[2] 该研究发现FXR是ACE2转录的直接调节因子。 (Z)-古古甾酮通过抑制FXR信号通路,降低多种与COVID-19相关的人类细胞类型(呼吸道、胆道、肠道)中ACE2的表达,从而降低体外细胞对SARS-CoV-2感染的易感性。[2] 其机制涉及(Z)-古古甾酮与FXR结合,阻止FXR与ACE2启动子上的FXR反应元件(IR-1)结合,导致ACE2转录减少。[2] 该研究表明,通过抑制FXR来调节ACE2等宿主受体,是一种潜在的针对宿主的COVID-19预防策略,与直接抗病毒策略相比,该策略可能更不易受到病毒逃逸突变的影响。[2] |
| 分子式 |
C21H28O2
|
|---|---|
| 分子量 |
312.45
|
| 精确质量 |
312.208
|
| 元素分析 |
C, 80.73; H, 9.03; O, 10.24
|
| CAS号 |
39025-23-5
|
| 相关CAS号 |
39025-23-5 (Z-Guggulsterone); 95975-55-6; 39025-24-6 (E-Guggulsterone)
|
| PubChem CID |
6450278
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.1±0.1 g/cm3
|
| 沸点 |
463.3±45.0 °C at 760 mmHg
|
| 熔点 |
188-190°
|
| 闪点 |
172.3±25.7 °C
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
| 折射率 |
1.557
|
| LogP |
3.65
|
| tPSA |
34.14
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
2
|
| 可旋转键数目(RBC) |
0
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
640
|
| 定义原子立体中心数目 |
5
|
| SMILES |
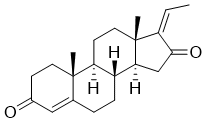
C/C=C/1\C(=O)C[C@H]2[C@@H]3CCC4=CC(=O)CC[C@]4(C)[C@H]3CC[C@]12C
|
| InChi Key |
WDXRGPWQVHZTQJ-OSJVMJFVSA-N
|
| InChi Code |
InChI=1S/C21H28O2/c1-4-16-19(23)12-18-15-6-5-13-11-14(22)7-9-20(13,2)17(15)8-10-21(16,18)3/h4,11,15,17-18H,5-10,12H2,1-3H3/b16-4+/t15-,17+,18+,20+,21-/m1/s1
|
| 化学名 |
(8R,9S,10R,13S,14S,17Z)-17-ethylidene-10,13-dimethyl-1,2,6,7,8,9,11,12,14,15-decahydrocyclopenta[a]phenanthrene-3,16-dione
|
| 别名 |
Z-Guggulsterone; (Z)-Guggulsterone; Z-Guggulsterone; Guggulsterone; 39025-23-5; 95975-55-6; Guggulsterones Z; Cis-Guggulsterone; Guggulsterone E&Z; (Z)-Guggulsterone
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 5~10 mg/mL (16.0~32.0 mM)
Ethanol: ~2 mg/mL (~6.4 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 1 mg/mL (3.20 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 10.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 2 中的溶解度: 10 mg/mL (32.01 mM) in 50% PEG300 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2005 mL | 16.0026 mL | 32.0051 mL | |
| 5 mM | 0.6401 mL | 3.2005 mL | 6.4010 mL | |
| 10 mM | 0.3201 mL | 1.6003 mL | 3.2005 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
|
 VEGFR2-IN-7
VEGFR2-IN-7
 SYHA1813
SYHA1813
 VEGFR-2-IN-38
VEGFR-2-IN-38
 BHEP
BHEP
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA




 463611831
463611831
