| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
Fexaramine (5, 25, and 50 μM) was used to treat bile acids to HuTu -80 cells for a duration of 24 hours. Assay values of the small heterodimer fraction (SHP) are increased by 2.1 times when fexaramine (50 μM) is added. Endogenous protein levels were dramatically decreased (33% drop in 50 μM Fexaramine) when cells were treated with varying doses of Fexaramine. Treatment with fexaramine nonetheless markedly reduced secretin promoter activity by 32% [1].
|
|---|---|
| 体外研究 (In Vitro) |
Fexaramine(5、25 和 50 μM)用于处理 HuTu -80 细胞的胆汁酸,持续时间为 24 小时。添加 fexaramine (50 μM) 后,小异二聚体组分 (SHP) 的测定值增加 2.1 倍。当细胞用不同剂量的 Fexaramine 处理时,内源蛋白水平显着降低(50 μM Fexaramine 下降 33%)。尽管如此,fexaramine 治疗仍显着降低了促胰液素启动子活性 32% [1]。
在基于FRET的共激活子结合实验中,Fexaramine 能诱导类固醇受体共激活子SRC-1肽段与FXR配体结合域(LBD)的结合,EC50为255 nM。该活性与合成激动剂GW4064(EC50 = 100 nM)相当。[2] 在CV-1细胞的瞬时转染实验中,当共转染FXR、RXR和含有多个FXR反应元件(FXRE)拷贝的报告基因时,Fexaramine(1 nM 至 1 µM)能以剂量依赖方式有效激活FXR:在ECRE元件上诱导100倍(1 µM时),在ER-8元件上诱导4倍(1 µM时)。[2] Fexaramine 在瞬时转染实验中也能够激活已知FXR靶基因的生理启动子。在1 µM浓度下,它诱导人回肠胆汁酸结合蛋白(I-BABP)启动子表达28倍,人磷脂转移蛋白(PLTP)启动子表达2-3倍,大鼠多药耐药相关蛋白2(MRP-2)启动子表达2-3倍。[2] 在使用嵌合核激素受体(NHR)构建体(GAL4 DBD融合各种NHR LBD)的交叉反应性研究中,Fexaramine(10 µM)对FXR显示出高选择性,且不激活其他NHR,包括RXRα、PPARα/γ/δ、PXR、LXRα、TRβ、RARβ、CAR、ERRγ或VDR。[2] 在稳定表达全长FXR的HT29人结肠癌细胞(HT29-FXRFL)中,用Fexaramine(1 nM 至 10 µM)处理能以剂量依赖方式诱导内源性I-BABP基因的表达,效果与GW4064相似。在缺乏功能性FXR的对照细胞系中未观察到诱导。[2] 在稳定表达FXR的HEPG2人肝癌细胞(HEPG2-FXR)中,用Fexaramine(10 nM 至 10 µM)处理可诱导内源性FXR靶基因(包括SHP、PLTP、BSEP和MRP-2)的表达,通过Northern blot检测。其诱导谱和效力与GW4064相当。[2] 用Fexaramine(10 µM)处理原代人肝细胞的基因表达谱分析显示,其基因组特征与天然胆汁酸CDCA(100 µM)或合成激动剂GW4064(10 µM)处理不同,表明配体对全局基因调控具有特异性影响。一部分基因受所有三种FXR激动剂的共同调控。[2] 解析了人FXR配体结合域与Fexaramine 复合物的高分辨率(1.78 Å)晶体结构。结构显示,Fexaramine 被包裹在一个726 ų的疏水腔中,与受体形成广泛的范德华接触和特定的氢键(涉及His298和Ser336),为其高亲和力和作用机制提供了结构基础。[2] |
| 体内研究 (In Vivo) |
使用 fexaramine 治疗的 DIO 小鼠产生了显着的代谢特征,包括炎症减轻、胰岛素肥胖增强、体重增加减少以及白色脂肪组织褐变 [3]。
在饮食诱导的肥胖小鼠中,长期口服Fexaramine(100毫克/公斤/天,持续5周)与溶剂对照组相比,显著减少了体重增加和脂肪量(皮下和内脏)。[3] Fexaramine治疗改善了DIO小鼠的代谢指标,包括降低血清葡萄糖、胰岛素、瘦素、胆固醇和抵抗素水平。[3] Fexaramine治疗降低了DIO小鼠血清中炎性细胞因子的水平。[3] Fexaramine治疗增强了DIO小鼠的葡萄糖耐受性和胰岛素敏感性,但对普通饲料喂养的小鼠无影响。[3] Fexaramine增加了DIO小鼠的能量消耗,表现为更高的耗氧量、二氧化碳产量、核心体温以及减少的棕色脂肪组织脂质积聚。[3] Fexaramine诱导了白色脂肪组织的褐变,表现为腹股沟白色脂肪组织中多房性、表达Ucp1的脂肪细胞增多,棕色脂肪样特征基因表达增加,以及腹股沟脂肪组织基质血管部分的呼吸能力增强。[3] Fexaramine改善了DIO小鼠的肝脏胰岛素敏感性,表现为高胰岛素-正葡萄糖钳夹实验中胰岛素介导的肝葡萄糖产生抑制显著增强。[3] Fexaramine改善了DIO小鼠的肝脏脂肪变性,表现为肝脏脂滴减少、肝脏甘油三酯含量降低以及肝脏糖异生和脂肪生成基因表达下调。[3] Fexaramine诱导的代谢改善在FXR敲除小鼠中被消除,证实了其FXR依赖性。[3] Fexaramine诱导了回肠中肠道激素Fgf15的表达并增加了循环中的Fgf15水平。[3] Fexaramine改变了胆汁酸组成,减少了胆汁酸池大小,降低了牛磺胆酸比例,增加了石胆酸比例,同时抑制了肝脏Cyp7a1表达并增加了Cyp7b1表达。[3] Fexaramine降低了肠道通透性并增加了粘膜防御基因的表达。[3] Fexaramine不激活G蛋白偶联胆汁酸受体Tgr5。在Gpbar1敲除小鼠中,部分代谢改善作用减弱,表明Tgr5通路参与了某些效应。[3] |
| 酶活实验 |
使用基于FRET的共激活子结合实验来测量激动剂诱导的FXR与其共激活子之间的相互作用。该实验使用了铕标记的GST-FXR-LBD融合蛋白和别藻蓝蛋白标记的含有共激活子SRC-1受体结合域(LXXLL基序)的肽段。将递增浓度的测试化合物加入到含有标记蛋白的反应混合物中。激动剂诱导的受体与肽段之间的接近增加了FRET信号,通过测量665 nm与615 nm处的发射比来量化。根据剂量反应曲线确定共激活子募集的EC50。[2]
|
| 细胞实验 |
瞬时转染报告基因实验: 将CV-1细胞接种于48孔板中,生长至60-70%汇合度。使用基于脂质的转染试剂共转染小鼠FXR和人RXRα的表达质粒、含有FXR反应元件(FXRE)或天然启动子(如I-BABP、PLTP、MRP-2)的荧光素酶报告质粒以及用于归一化的对照质粒(如pCMX-LacZ)。24小时后更换培养基,并用溶解于DMSO中的递增浓度测试化合物处理细胞。转染后36-48小时收集细胞。测量荧光素酶活性,并归一化至对照质粒的β-半乳糖苷酶活性。[2]
稳定细胞系基因诱导实验: 通过逆转录病毒转导和筛选,建立了稳定表达FXR(或对照构建体)的HT29结肠癌细胞或HEPG2肝癌细胞。对于诱导实验,细胞在用活性炭剥离血清的培养基中培养至汇合,处理前24小时更换为含药培养基。然后用指定浓度的化合物(如Fexaramine、GW4064、CDCA)或载体(DMSO)处理24-48小时。使用酚-氯仿类试剂分离总RNA。对于Northern印迹分析,每个样品取20 µg总RNA在变性琼脂糖-甲醛凝胶上电泳,转移至尼龙膜并UV交联。制备针对靶基因(如I-BABP、SHP、PLTP、BSEP、MRP-2)和上样对照(如β-actin、36B4)的放射性标记cDNA探针并与膜杂交。通过放射自显影观察基因表达水平,并使用磷屏成像仪进行定量。[2] 嵌合NHR选择性实验: 将CV-1细胞瞬时转染一种嵌合表达质粒,其中酵母GAL4 DNA结合域与各种核受体(FXR、RXRα、PPARs等)的配体结合域融合。共转染一个含有最小胸苷激酶启动子、上游有四个GAL4结合位点和荧光素酶基因的报告质粒。转染后,用DMSO或10 µM测试化合物处理细胞。测量荧光素酶活性并归一化至内参(如β-半乳糖苷酶),数据以相对于未处理细胞的激活倍数表示。[2] |
| 动物实验 |
对于DIO小鼠的慢性治疗:野生型雄性C57BL/6J小鼠从6周龄开始喂食60%高脂饮食(HFD),持续14周以诱导肥胖。随后,在HFD喂养期间,小鼠每日灌胃给予Fexaramine(50或100 mg/kg体重)或载体(玉米油),持续5周。[3]
对于急性靶基因诱导:小鼠每日灌胃或腹腔注射Fexaramine(100 mg/kg)或载体,持续3-5天,并在末次给药后1小时收集组织进行基因表达分析。[3] 对于高胰岛素-正常血糖钳夹试验:DIO小鼠(HFD喂养12周)在钳夹试验前3周每日灌胃给予Fexaramine(100 mg/kg)或载体。在颈静脉植入双导管,待小鼠恢复并禁食后,开始持续输注D-[3-3H]葡萄糖。示踪剂达到平衡后,开始输注胰岛素和不同剂量的葡萄糖以维持血糖正常,并采集血样以计算肝糖生成率和葡萄糖清除率。[3] 代谢表型分析:将小鼠置于综合实验动物监测系统(CLAMS)中,连续数日测量其二氧化碳生成量、氧气消耗量、呼吸商和活动计数。[3] 肠道通透性评估:小鼠口服异硫氰酸荧光素(FITC)-葡聚糖,并在特定时间后测量血清荧光值以评估肠道渗漏情况。[3] |
| 药代性质 (ADME/PK) |
口服(100 mg/kg)后,非萨拉明的全身吸收极少。与腹腔注射相比,口服给药后的血清药物浓度低一个数量级。[3]
口服后非萨拉明的血清浓度低于其25 nM的半数最大有效浓度(EC50),这与其肠道限制性作用以及在肝肾中缺乏靶基因激活相符。[3] 该论文未提供非萨拉明的详细药代动力学参数,例如半衰期、口服生物利用度、清除率或分布容积。[3] |
| 毒性/毒理 (Toxicokinetics/TK) |
在 DIO 小鼠中,长期口服 Fexaramine(100 mg/kg/天,持续 5 周)未观察到肠道毒性迹象。[3]
Fexaramine 治疗降低了 DIO 小鼠的血清丙氨酸氨基转移酶 (ALT) 水平,表明肝损伤减轻。[3] |
| 参考文献 |
|
| 其他信息 |
非索拉明属于联苯类化合物。
非索拉明是一种合成的非甾体类高亲和力法尼醇X受体 (FXR) 激动剂,它是通过筛选和优化基于苯并吡喃的组合化学库而发现的。其化学结构与天然胆汁酸配体(例如CDCA)和其他合成FXR激动剂(例如GW4064)均不同。[2] FXR的主要功能是作为胆汁酸传感器,协调胆固醇代谢、脂质稳态以及膳食脂肪/维生素的吸收。非索拉明是一种有价值的化学工具,可用于解析FXR特异性通路,从而将其与天然胆汁酸更广泛的生物学效应区分开来,因为天然胆汁酸可以通过多种受体和信号通路发挥作用。 [2] FXR 与 Fexaramine 结合的高分辨率晶体结构为 FXR 配体结合口袋的结构提供了关键信息,并作为模拟天然胆汁酸结合的模板,解释了不同胆汁酸衍生物的构效关系。[2] 基因表达谱分析表明,不同类型的 FXR 激动剂(Fexaramine、GW4064、CDCA)可以产生不同的转录谱,提示可能存在配体特异性效应,并凸显了 Fexaramine 等特异性合成探针在研究 FXR 基因网络方面的实用性。[2] |
| 分子式 |
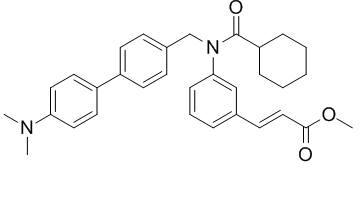
C32H36N2O3
|
|---|---|
| 分子量 |
496.6398
|
| 精确质量 |
496.273
|
| CAS号 |
574013-66-4
|
| PubChem CID |
5326713
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| 密度 |
1.158g/cm3
|
| 沸点 |
677.7ºC at 760 mmHg
|
| 闪点 |
363.7ºC
|
| 折射率 |
1.626
|
| LogP |
6.719
|
| tPSA |
49.85
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
37
|
| 分子复杂度/Complexity |
743
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CN(C)C1=CC=C(C=C1)C2=CC=C(C=C2)CN(C3=CC=CC(=C3)/C=C/C(=O)OC)C(=O)C4CCCCC4
|
| InChi Key |
VLQTUNDJHLEFEQ-KGENOOAVSA-N
|
| InChi Code |
InChI=1S/C32H36N2O3/c1-33(2)29-19-17-27(18-20-29)26-15-12-25(13-16-26)23-34(32(36)28-9-5-4-6-10-28)30-11-7-8-24(22-30)14-21-31(35)37-3/h7-8,11-22,28H,4-6,9-10,23H2,1-3H3/b21-14+
|
| 化学名 |
methyl (E)-3-(3-(N-((4'-(dimethylamino)-[1,1'-biphenyl]-4-yl)methyl)cyclohexanecarboxamido)phenyl)acrylate
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~50 mg/mL (~100.68 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 2.75 mg/mL (5.54 mM) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
*生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.03 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.03 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0135 mL | 10.0677 mL | 20.1353 mL | |
| 5 mM | 0.4027 mL | 2.0135 mL | 4.0271 mL | |
| 10 mM | 0.2014 mL | 1.0068 mL | 2.0135 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
