| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
Syk (IC50 = 41 nM)
R406, a spleen tyrosine kinase (Syk) inhibitor, is prodrugged by R788. R788 has a Ki of 30 nM and functions as a competitive inhibitor of ATP binding. With an EC50 of 56 nM, R788 dose-dependently prevents anti-IgE-mediated CHMC degranulation. R788 also prevents the release of LTC4 and cytokines and chemokines, such as TNFα, IL-8, and GM-CSF, that are triggered by anti-IgE. All phosphorylation events downstream of Syk signaling are inhibited when R788 inhibits Syk. The most potent inhibitor of IL-4 and IL-2 receptor signaling in CHMC is R788, followed by FcρRI signaling. R788 specifically inhibits human mast cells, neutrophils, and macrophages' FcγR signaling. R788 has the ability to prevent immune complex-mediated local inflammatory injury.R788 causes most of the tested DLBCL cell lines to undergo apoptosis[1]. R788 selectively inhibits both tonic- and ligand-induced BCR signaling (autophosphorylation of SYK525/526 and SYK-dependent phosphorylation of the B-cell linker protein [BLNK]) in R788-sensitive DLBCL cell lines[2]. |
|---|---|
| 体外研究 (In Vitro) |
体外活性:R935788是R406的亚甲基磷酸酯前药,在体内可快速转化为R406。 R406(R935788 的体外活性形式)选择性抑制 Syk 依赖性信号传导,EC50 值范围为 33 nM 至 171 nM,比不同细胞中的 Syk 独立途径更有效。 R406 抑制多种弥漫性大 B 细胞淋巴瘤 (DLBCL) 细胞系的细胞增殖,EC50 值范围为 0.8 μM 至 8.1 μM。 R406 治疗不仅在高 (TCL-002) 细胞中而且在磷酸化 Syk 水平低的细胞中降低 BLNK、Akt、糖原合酶激酶 3 (GSK-3)、叉头盒 O (FOXO) 和 ERK 的基础磷酸化(TCL1-551)。此外,R406 完全抑制 TCL1 白血病中抗 IgM 诱导的 Bcr 信号。尽管 TCL1 白血病中的组成型活性 Syk 水平较高,但 R406 对白血病细胞没有选择性细胞毒性。激酶测定:R406(R935788 的体外活性形式)在 DMSO 中连续稀释,然后在激酶缓冲液(20 mM HEPES,pH 7.4,5 mM MgCl2,2 mM MnCl2,1 mM DTT,0.1 mg/)中稀释至 1% DMSO mL 乙酰化 BGG)。在室温下添加激酶缓冲液中的 ATP 和底物,最终 DMSO 浓度为 0.2%。激酶反应在含有 5 μM HS1 肽底物和 4 μM ATP 的最终体积 20 μL 中进行,并通过在激酶缓冲液中添加 0.125 ng Syk 开始。使反应在室温下进行40分钟。通过添加 20 μL PTK 淬灭混合物来终止反应,该混合物含有用 FP 稀释缓冲液稀释的 EDTA/抗磷酸酪氨酸抗体(1× 最终)/荧光磷酸肽示踪剂(0.5× 最终)。将板在室温下黑暗中孵育 30 分钟,然后在 Polarion 荧光偏振板读数器上读数。使用通过与酪氨酸激酶测定试剂盒中提供的磷酸肽竞争剂竞争生成的校准曲线来转换数据以确定存在的磷酸肽的量。对于 IC50 测定,R406 在 11 个浓度下进行重复测试,并使用 Prism GraphPad 软件通过非线性回归分析进行曲线拟合。细胞测定:将细胞(TCL1-002、TCL1-252、TCL1-551、TCL1-870 和 TCL1-540)暴露于浓度不断增加的 R406(R935788 的体外活性形式)中 48 小时。通过碘化丙啶 (PI) 和膜联蛋白-A5-FITC 缀合物双重染色测定凋亡细胞的百分比。 Ki-67 染色使用 FITC 小鼠抗 Ki-67 套件进行。使用 CellQuest 3.3 版软件在 FACSCalibur 流式细胞仪上分析样品。
R406 剂量依赖性地抑制抗IgE介导的培养人肥大细胞脱颗粒(类胰蛋白酶释放),EC₅₀ 为 0.056 ± 0.02 µM,但对离子霉素触发的脱颗粒无活性,表明其对FcεRI信号传导具有特异性。 [1] R406 抑制抗IgE诱导的CHMC中LTC₄ (EC₅₀ = 0.093 µM)、TNFα (EC₅₀ = 0.118 µM)、IL-8 (EC₅₀ = 0.140 µM) 和 GM-CSF (EC₅₀ = 0.158 µM) 的产生和释放。 [1] R406 也抑制抗IgG诱导的CHMC脱颗粒 (EC₅₀ = 0.064 µM) 和介质释放(LTC₄、TNFα、IL-8、GM-CSF),表明其抑制FcγR信号传导。 [1] R406 抑制人单核细胞来源的巨噬细胞 (EC₅₀ = 0.111 µM) 和单核细胞系THP-1 (EC₅₀ = 0.171 µM) 中由FcγR交联诱导的TNFα产生。 [1] R406 抑制经TNFα预处理的、并用抗IgG刺激的人中性粒细胞的氧化爆发 (EC₅₀ = 0.033 µM)。 [1] R406 抑制用抗IgM刺激的原代人B细胞中CD69的上调 (EC₅₀ = 0.048 µM),表明其抑制B细胞受体信号传导。 [1] R406 不抑制CHMC中Syk酪氨酸352或FcRγ链的磷酸化,但抑制T细胞连接蛋白(LAT)酪氨酸191以及下游信号分子(PLCγ1、Akt、ERK、p38、JNK)的磷酸化,证实了其对Syk的选择性抑制。 [1] 在细胞选择性评估中,R406 抑制Flt3自身磷酸化的效力比抑制Syk低约5倍,并对Jak和Lck依赖性途径显示出较弱的抑制。 [1] R406 (高达50 µM) 对全血中单核细胞和粒细胞对FITC标记的调理细菌的吞噬作用影响可忽略不计。 [1] R406 不显著抑制由调理大肠杆菌吞噬作用诱导的人白细胞氧化爆发活性,在10 µM浓度下也不影响粒细胞向fMLP的迁移。 [1] R406 (高达20 µM) 不抑制纯化的原代中性粒细胞对金黄色葡萄球菌的杀菌活性。 [1] |
| 体内研究 (In Vivo) |
鉴于 R406 在小鼠体内的血浆半衰期小于 2 小时,R935788 以 3 小时的间隔分 3 次给药,以在每天的治疗期间提供持续的 Syk 抑制,模仿人类较长的血浆半衰期(15小时)。尽管体外细胞毒性作用相对较小,但 R935788 在体内显着抑制白血病细胞的增殖和存活,这与阻断抗原依赖性 B 细胞受体 (Bcr) 信号传导有关,而不是抑制组成型 Syk 活性。 R935788 按 80 mg/kg/天治疗 18-21 天,可有效抑制在治疗最后一天白血病 CD5+/B220+ 细胞无法检测到的小鼠中 TCL1-002、TCL1-551 和 TCL1-870 的肿瘤生长,显着延长生存期接受治疗的小鼠的中位生存期从45/46天增加到170/172天,并且在6个月的随访期后完全消除了相当比例的小鼠中的恶性细胞,且不影响正常B淋巴细胞的产生。 R935788 治疗还诱导正常和恶性 B 细胞从脾和淋巴结早期短暂迁移到外周血,随后选择性抑制恶性 B 细胞群的生长。此外,R935788还可以有效对抗Eμ-TCL1转基因小鼠自发发生的TCL1白血病。
口服给予 R406 (1和5 mg/kg,攻击前1小时) 分别使小鼠皮肤反向被动Arthus反应减少约72%和86%(通过伊文思蓝染料外渗测量)。 [1] 在胶原抗体诱导的关节炎小鼠模型中,口服给予 R406 (抗体攻击后4小时开始,每日两次) 减少了爪部炎症和肿胀,减缓了疾病进展,并改善了关节组织病理学(减少滑膜炎、血管翳形成、白细胞浸润)。 [1] 在K/BxN血清转移小鼠模型中,口服给予 R406 (10 mg/kg,每日两次) 延迟了临床关节炎的发作并降低了其严重程度,爪部增厚和临床评分减少了约50%。 [1] 口服高达100 mg/kg的 R406 (导致全身暴露高达25 µM) 在小鼠尾尖截断后并未延长出血时间。 [1] |
| 酶活实验 |
有荧光偏振反应。为了确定 Ki,在重复的 200 μL 反应中设置八个不同的 ATP 浓度,从 200 μM(2 倍连续稀释)开始,有或没有 DMSO 或 R788,浓度为 125、62.5、31.25、15.5 或 7.8 nM 。取出每个反应20微升并在不同时间淬灭以结束反应。将反应速率与 ATP 浓度作图,得出每个 R788 浓度的表观 Km 和 Vmax。为了计算 Ki,最终根据抑制剂浓度绘制表观 Km(或表观 Ki/Vmax)。
荧光偏振激酶实验: 在存在DMSO或浓度为125、62.5、31.25、15.5或7.8 nM的 R406 的情况下,在八种不同ATP浓度(从200 µM开始进行系列稀释)下设置重复的200 µl反应体系。在不同时间点,取出20 µl各反应液并淬灭。测定每个ATP浓度下的反应速率,并绘制相对于ATP浓度的曲线以确定表观Km和Vmax。然后将表观Km相对于抑制剂浓度作图以确定Ki。使用酶动力学软件进行数据分析。 [1] 晶体学实验: Syk激酶结构域(Ile365至Asn635,带有R440Q突变)被结晶。衍射数据收集至2.3-Å分辨率。使用先前确定的Syk激酶结构作为搜索模型,通过分子置换法解析结构。将晶体与 R406 共浸泡,显示的电子密度与 R406 竞争性结合于ATP结合口袋一致。 [1] |
| 细胞实验 |
补充数据显示了培养的人类肥大细胞 (CHMC) 的生长、启动和刺激,这些细胞源自脐带血 CD34+ 祖细胞。刺激前将细胞在 DMSO 或 R788 中培养半小时。下一步涉及使用 2 μM 离子霉素或 0.25 至 2 mg/mL 抗 IgE 或抗 IgG 刺激细胞。使用改良的 Tyrode 缓冲液刺激每孔约 1500 个细胞 30 分钟,以测量类胰蛋白酶。每孔一万个细胞被刺激一小时以产生 LTC4,刺激七小时以产生细胞因子。 LTC4 和细胞因子使用 Luminex 多重技术进行检测,而类胰蛋白酶活性则通过肽底物的发光读数来确定。
人肥大细胞脱颗粒和介质释放: 源自脐带血CD34⁺祖细胞的培养人肥大细胞经预处理,并与 R406 或DMSO预孵育30分钟。用抗IgE或抗IgG刺激细胞。30分钟后使用发光肽底物法测定类胰蛋白酶释放。对于LTC₄和细胞因子(TNFα、IL-8、GM-CSF)的产生,将细胞刺激1或7小时,并使用多重技术测量介质。 [1] 信号传导分析的Western Blot: 将CHMC或其他细胞类型与 R406 或对照抑制剂(如PP2)预孵育40分钟,并用抗IgE刺激5分钟。细胞在SDS样品缓冲液中裂解。蛋白质通过SDS-PAGE分离,转移至膜上,并用针对Syk (Tyr352)、LAT (Tyr191)、PLCγ1 (Tyr783)、Akt (Ser473)、ERK (Thr202/Tyr204)、p38 (Thr180/Tyr182) 和JNK (Thr183/Tyr185) 的磷酸化特异性抗体进行检测。对膜进行总蛋白复染以验证上样量均等。对于免疫沉淀,刺激并裂解细胞,然后使用特异性抗体按照标准免疫沉淀方案进行。 [1] 巨噬细胞TNFα产生: 将源自CD14⁺单核细胞(用GM-CSF分化5天)的人原代巨噬细胞或用IFN-γ预处理的THP-1细胞,在存在 R406 的情况下加入IgG包被的孔中。孵育16-20小时后,通过多重测定法测量上清液中的TNFα。在未包被的孔中用LPS刺激作为Syk非依赖性对照。 [1] 中性粒细胞氧化爆发: 分离原代人中性粒细胞,在缓冲液中与 R406 预孵育,用TNFα预处理,并用抗IgG或PMA刺激。通过加入二氢罗丹明123并用流式细胞术分析细胞内荧光来测量呼吸爆发。 [1] B细胞活化: 从外周血中分离的原代人B细胞在培养基中与 R406 预孵育60分钟,用抗IgM刺激6小时,用抗CD69-APC染色,并通过流式细胞术分析。 [1] |
| 动物实验 |
患有关节炎的 Balb/c 小鼠
1 mg/kg 或 5 mg/kg og 反向被动 Arthus 反应:雌性 C57BL/6 小鼠经静脉注射 1% 卵清蛋白 (OVA) 和 1% 伊文思蓝染料 (10 mg/kg) 进行激发。10 分钟后,在小鼠背部一侧的三个部位皮内注射兔抗 OVA IgG(50 µg,25 µl);在另一侧注射对照兔 IgG。R406 或载体(67% PEG 400)于激发前 60 分钟口服给药。4 小时后,处死小鼠,取注射部位 8 mm 的穿刺活检组织,在 80°C 的甲酰胺中孵育过夜。用分光光度计在 OD610 处测定外渗染料浓度。 [1]胶原抗体诱导关节炎 (CAIA):雌性 Balb/c 小鼠在第 0 天通过静脉注射胶原诱导关节炎单克隆抗体混合物进行被动致敏,随后在第 2 天腹腔注射 LPS (25 µg)。R406 或载体(35% TPGS、60% PEG 400、5% 丙二醇)于第 0 天抗体激发后 4 小时开始,每日两次口服给药,持续 14 天。定期评估临床评分。 [1] K/BxN 血清转移模型:在第 0 天和第 2 天,通过腹腔注射 150 µl K/BxN 小鼠混合血清,在 C57BL/6 小鼠中诱导关节炎。R406 或载体(35% TPGS、60% PEG 400、5% 丙二醇)在血清注射前 1 小时口服给药,之后每日两次,持续 13 天。每日评估踝关节厚度和临床关节炎评分。[1] 出血时间测定:小鼠口服 R406(最高剂量 100 mg/kg)或阿司匹林(100 mg/kg)。尾尖切除后测量出血时间。[1] |
| 药代性质 (ADME/PK) |
在一项针对健康志愿者的临床研究中,R406 具有很高的口服生物利用度。血浆浓度随剂量呈比例增加,直至 400 mg,之后达到平台期。[1] 血浆浓度峰值通常在给药后 1.2 至 1.3 小时之间达到。[1] 末端半衰期约为 15 小时。[1] R406 在人体内表现出很高的血浆蛋白结合率(>98%)。[1] 体外实验中,使嗜碱性粒细胞活化(CD63 表达)降低 50% 的血浆浓度为 496 ± 42 ng/ml,对应的 EC₅₀ 为 1.06 µM。[1]
|
| 毒性/毒理 (Toxicokinetics/TK) |
妊娠期和哺乳期影响
◉ 哺乳期用药概述 目前尚无关于福斯他替尼在哺乳期临床应用的信息。由于福斯他替尼的活性代谢物(R406)与血浆蛋白的结合率高达98.3%,因此其在乳汁中的含量可能很低。然而,该活性代谢物的半衰期为15小时,可能会在婴儿体内蓄积。制造商建议在接受福斯他替尼治疗期间以及末次给药后至少1个月内停止母乳喂养。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对泌乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 在细胞选择性分析中,R406在生化结合试验中显示出对腺苷A₃受体(IC₅₀ ≈ 0.081 µM)、腺苷转运体(IC₅₀ ≈ 1.84 µM)和单胺转运体(IC₅₀ ≈ 2.74 µM)的抑制作用。 [1] 在人体志愿者中,即使在最高剂量(高达 600 mg)下,R406 也未抑制富血小板血浆中胶原蛋白或 ADP 诱导的血小板聚集。[1] 在人体志愿者中进行的安全性评估表明,在抑制嗜碱性粒细胞活化的剂量下,R406 对血液学或化学安全参数无影响。[1] 在 600 mg 剂量组中最常见的不良事件是体位性眩晕(6 名受试者中有 5 名)。[1] 在人体中,R406 治疗导致循环 CD45+CD14+ 单核细胞在单次给药后 4 小时呈剂量依赖性减少,并在 20 小时后恢复正常。[1] |
| 参考文献 | |
| 其他信息 |
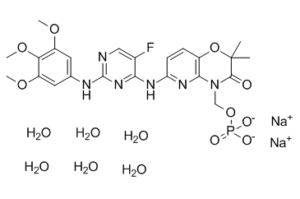
福斯他替尼二钠是Syk激酶抑制剂福斯他替尼的口服二钠盐,具有潜在的抗炎和免疫调节活性。福斯他替尼抑制Syk激酶介导的IgG Fcγ受体信号传导,从而抑制肥大细胞、巨噬细胞和B细胞的活化以及相关的炎症反应和组织损伤。 Syk激酶广泛表达于造血细胞中,是一种非受体酪氨酸激酶,参与将活化的免疫受体与下游信号事件偶联,从而介导多种细胞反应,包括增殖、分化和吞噬作用。
另见:福斯他替尼二钠(注释已移至)。 药物适应症 Tavlesse®适用于治疗对其他治疗无效的成人慢性免疫性血小板减少症 (ITP)。 R406是一种小分子,化学名称为N4-(2,2-二甲基-3-氧代-4H-吡啶并[1,4]恶嗪-6-基)-5-氟-N2-(3,4,5-三甲氧基苯基)-2,4-嘧啶二胺。 [1] 该化合物是通过利用人肥大细胞进行基于细胞的构效关系筛选,以寻找FcεRI信号通路抑制剂而发现的。[1] R406以U形构象结合Syk的ATP结合口袋,其关键相互作用位于铰链区,通过嘧啶N1和连接基N2与Syk结合,此外还与甲氧基化的苯环和杂环形成氢键和疏水相互作用。[1] R406抑制免疫细胞(肥大细胞、巨噬细胞、中性粒细胞、B细胞)中Syk依赖性信号通路,而不会显著干扰小鼠体内Syk非依赖性先天免疫反应(吞噬作用、杀菌活性)或止血功能。[1] 该药物在类风湿性关节炎等免疫复合物介导的炎症性疾病动物模型中显示出治疗潜力。 [1] 在人体中,口服R406可达到抑制Syk依赖性IgE介导的嗜碱性粒细胞活化的药物浓度,提示其可能具有调节人类过敏和自身免疫性疾病中Syk活性的潜力。[1] |
| 分子式 |
C₂₃H₂₄FN₆NA₂O₉P.₆H₂O
|
|
|---|---|---|
| 分子量 |
732.51
|
|
| 精确质量 |
732.18
|
|
| 元素分析 |
C, 37.71; H, 4.95; F, 2.59; N, 11.47; Na, 6.28; O, 32.76; P, 4.23
|
|
| CAS号 |
914295-16-2
|
|
| 相关CAS号 |
Fostamatinib Disodium;1025687-58-4;Fostamatinib;901119-35-5
|
|
| PubChem CID |
24828759
|
|
| 外观&性状 |
White to gray solid powder
|
|
| LogP |
3.241
|
|
| tPSA |
205.76
|
|
| 氢键供体(HBD)数目 |
8
|
|
| 氢键受体(HBA)数目 |
21
|
|
| 可旋转键数目(RBC) |
9
|
|
| 重原子数目 |
48
|
|
| 分子复杂度/Complexity |
893
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
[Na].O=C1C(C)(C)OC2C(=NC(NC3C(F)=CN=C(NC4C=C(OC)C(OC)=C(OC)C=4)N=3)=CC=2)N1COP(O)(O)=O.O.[Na].O.O.O.O.O
|
|
| InChi Key |
ZQGJCHHKJNSPMS-UHFFFAOYSA-L
|
|
| InChi Code |
InChI=1S/C23H26FN6O9P.2Na.6H2O/c1-23(2)21(31)30(11-38-40(32,33)34)20-14(39-23)6-7-17(28-20)27-19-13(24)10-25-22(29-19)26-12-8-15(35-3)18(37-5)16(9-12)36-4;;;;;;;;/h6-10H,11H2,1-5H3,(H2,32,33,34)(H2,25,26,27,28,29);;;6*1H2/q;2*+1;;;;;;/p-2
|
|
| 化学名 |
disodium;[6-[[5-fluoro-2-(3,4,5-trimethoxyanilino)pyrimidin-4-yl]amino]-2,2-dimethyl-3-oxopyrido[3,2-b][1,4]oxazin-4-yl]methyl phosphate;hexahydrate
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: 8.33 mg/mL (11.37 mM) in 0.5% CMC-Na/saline water (这些助溶剂从左到右依次添加,逐一添加), 悬浮液;超声助溶。
*生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 10 mg/mL (13.65 mM) in Cremophor EL (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 View More
配方 3 中的溶解度: 0.5% CMC+0.25% Tween 80,pH6.5: 30 mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.3652 mL | 6.8258 mL | 13.6517 mL | |
| 5 mM | 0.2730 mL | 1.3652 mL | 2.7303 mL | |
| 10 mM | 0.1365 mL | 0.6826 mL | 1.3652 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT02077192
Conditions:Immune Thrombocytopenic PurpuraLink: https://clinicaltrials.gov/ct2/show/NCT03764618
Conditions:Warm Antibody Autoimmune Hemolytic AnemiaLink: https://clinicaltrials.gov/ct2/show/NCT04138927
Conditions:Warm Antibody Autoimmune Hemolytic Anemia
Title:A Efficacy and Safety Study of R935788 in the Treatment of Persistent/Chronic Immune Thrombocytopenic Purpura (ITP)
Status:Completed
updateDate:2019-02-12
Ctid:NCT02076399
Link: https://clinicaltrials.gov/ct2/show/NCT02076399
Conditions:Immune Thrombocytopenic PurpuraLink: https://clinicaltrials.gov/ct2/show/NCT02076412
Conditions:Immune Thrombocytopenic PurpuraLink: https://clinicaltrials.gov/ct2/show/NCT03363334
Conditions:Immune Thrombocytopenic PurpuraLink: https://clinicaltrials.gov/ct2/show/NCT00665626
Conditions:Rheumatoid ArthritisLink: https://clinicaltrials.gov/ct2/show/NCT00798096
Conditions:T Cell LymphomaLink: https://clinicaltrials.gov/ct2/show/NCT00665925
Conditions:Rheumatoid ArthritisLink: https://clinicaltrials.gov/ct2/show/NCT02433236
Conditions:IGA NephropathyLink: https://clinicaltrials.gov/ct2/show/NCT00923481
Conditions:Head and Neck Neoplasms|Pheochromocytoma|Colorectal Neoplasms|Carcinoma, Non-Small-Cell Lung|Carcinoma, Renal CellLink: https://clinicaltrials.gov/ct2/show/NCT00805467
Conditions:Rheumatoid ArthritisLink: https://clinicaltrials.gov/ct2/show/NCT00752999
Conditions:Systemic Lupus ErythematosusLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2013-005452-15
Condition:Persistent/Chronic Immune Thrombocytopenic PurpuraLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2013-005453-76
Condition:Persistent/Chronic Immune Thrombocytopenic PurpuraPerzistentní chronická imunitní trombocytopenická purpuraLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2014-000331-16
Condition:IgA nephropathy (IgAN)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2011-006070-73
Condition:Rheumatoid arthritisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2011-005371-16
Condition:Diffuse Large B-cell LymphomaLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-023692-26
Condition:Rheumatoid arthritisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-020892-22
Condition:Rheumatoid ArthritisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-020745-27
Condition:Rheumatoid ArthritisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-020744-35
Condition:Rheumatoid ArthritisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2010-020743-12
Condition:Rheumatoid arthritisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-009034-32
Condition:Chronic Lymphocytic leukemiaLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2008-000742-30
Condition:Rheumatoid ArthritisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2008-000743-34
Condition:Rheumatoid arthritisLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2008-000744-13
Condition:Rheumatoid Arthritis |
|---|
 |
 |
 Syk Kinase Peptide Substrate, Biotin labeled
Syk Kinase Peptide Substrate, Biotin labeled
 Syk-IN-7
Syk-IN-7
 Syk-IN-4
Syk-IN-4
 Dehydroabietinol
Dehydroabietinol
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
