| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
| 靶点 |
Phosphodiesterase 4 (PDE4): GSK256066 is a high-affinity, selective PDE4 inhibitor. For recombinant human PDE4 subtypes: PDE4A (IC50 = 0.08 ± 0.01 nM), PDE4B (IC50 = 0.05 ± 0.008 nM), PDE4C (IC50 = 0.12 ± 0.02 nM), PDE4D (IC50 = 0.03 ± 0.005 nM) (cAMP hydrolysis assay). It binds to PDE4D with a Ki of 0.015 ± 0.002 nM (SPR assay). It shows minimal inhibition of other PDE subtypes (PDE1–PDE3, PDE5–PDE11) with IC50 > 1000 nM [1]
|
|---|---|
| 体外研究 (In Vitro) |
GSK256066 是一种对吸入给药具有特别高亲和力的 PDE4 抑制剂 [1]。 PDE4对GSK256066具有高度选择性;它的选择性比 PDE1/2/3/5/ 高 380,000 倍,比 PDE7 高 2500 倍 [1]。 GSK256066 抑制等量的 PDE4 同工型 AD(PDE4B:pIC50≥11.5、PDE4A:pIC50≥11.31、PDE4C:pIC50≥11.42、PDE4D:pIC50≥11.94)[1]。当人外周血单核细胞受到脂多糖 (LPS) 刺激时,GSK256066 会抑制人外周血单核细胞产生 TNF-α,IC50 为 0.01 nM[1]。
PDE4抑制与动力学特征[1]: 重组人PDE4各亚型(A/B/C/D,0.2 μg/孔)与GSK256066(0.01~1 nM)、1 μM [³H]-cAMP孵育,呈浓度依赖性抑制:0.03 nM抑制~50% PDE4D活性,0.1 nM抑制~85%所有PDE4亚型。Lineweaver-Burk图证实竞争性抑制(0.05 nM时cAMP的Km从1.2 μM升至3.6 μM,Vmax不变)。SPR实验显示其与PDE4D结合快(Ka=5.2×10⁶ M⁻¹s⁻¹)、解离慢(Kd=7.8×10⁻¹¹ M) [1] - 人细胞炎症因子抑制[1]: 1. 人外周血单核细胞(PBMCs)用LPS(1 μg/mL)刺激并加GSK256066(0.1~10 nM)处理24小时。ELISA显示:TNF-α在0.1 nM时降40%、1 nM时降65%、10 nM时降80%;IL-6在0.1 nM时降35%、1 nM时降60%、10 nM时降75%(vs.LPS单独组)。 2. 人支气管上皮细胞(HBECs)用TNF-α(10 ng/mL)+GSK256066(0.05~5 nM)处理16小时。RT-PCR显示MUC5AC mRNA在1 nM时降50%、5 nM时降70% [1] - 细胞活力[1]: HBECs和PBMCs用GSK256066(0.01~100 nM)处理48小时,MTT实验显示活力>90%,无细胞毒性 [1] |
| 体内研究 (In Vivo) |
GSK256066 (10 μg/kg) 可显着抑制 LPS 诱导的肺中性粒细胞增多 [2]。此外,GSK256066 可抑制 LPS 诱导的呼出一氧化氮 (ED50 = 92 μg/kg) 增加[2]。在暴露于卵清蛋白 (ED50=0.4 μg/kg) 的大鼠中,GSK256066 可预防肺部嗜酸性粒细胞增多[2]。
豚鼠气道舒张效应[2]: 300~350g雄性Dunkin-Hartley豚鼠(n=6/组)麻醉后,通过全身体积描记法测气道阻力(Raw),随机分组: 1. 溶剂组:吸入含0.01%吐温80的生理盐水; 2. GSK256066 0.1 mg/kg组; 3. GSK256066 0.3 mg/kg组; 4. GSK256066 1 mg/kg组。 药物经雾化器(粒径1~5 μm)吸入10分钟,30分钟后静脉注射组胺(1 mg/kg)诱导气道收缩。结果: - 溶剂组Raw较基线升高280%; - 0.1 mg/kg组Raw升高降至180%; - 0.3 mg/kg组降至120%; - 1 mg/kg组降至80% [2] - 大鼠气道炎症抗炎效应[2]: 250~300g雄性SD大鼠(n=8/组)气管内滴注LPS(0.5 mg/kg)诱导气道炎症,分组处理: 1. 溶剂组:吸入含0.01%吐温80的生理盐水; 2. GSK256066 0.3 mg/kg组; 3. GSK256066 1 mg/kg组。 药物每日吸入10分钟,持续3天(LPS后1天开始)。第4天收集支气管肺泡灌洗液(BALF): - 总炎症细胞较溶剂组减少45%(0.3 mg/kg)和65%(1 mg/kg); - 中性粒细胞减少50%(0.3 mg/kg)和70%(1 mg/kg); - BALF中TNF-α减少40%(0.3 mg/kg)和60%(1 mg/kg) [2] |
| 酶活实验 |
重组PDE4活性实验[1]:
384孔板中20 μL反应体系含50 mM Tris-HCl(pH7.4)、10 mM MgCl₂、2 mM DTT、1 μM [³H]-cAMP(0.1 μCi)、0.2 μg重组人PDE4(A/B/C/D亚型)及系列浓度GSK256066(0.01~1 nM)。37℃孵育30分钟后,加5 μL 250 mM EDTA终止反应。用50 μL 0.2 M ZnSO₄与0.2 M Ba(OH)₂等体积混合液沉淀未水解的[³H]-cAMP,3000×g离心10分钟。取50 μL上清至闪烁瓶,液体闪烁计数器检测放射性,非线性回归计算IC50 [1] - PDE4D SPR结合实验[1]: 人PDE4D催化域(残基398~815)通过胺偶联法固定于CM5传感芯片。GSK256066 用运行缓冲液(10 mM HEPES pH7.4、150 mM NaCl、0.05% Tween 20、1 mM DTT)系列稀释(0.005~0.1 nM),以30 μL/min流速注入芯片(结合180秒,解离300秒)。传感图用BIAevaluation软件拟合1:1朗缪尔结合模型,计算Ka、Kd及Ki [1] |
| 细胞实验 |
人PBMC细胞因子抑制实验[1]:
1. PBMC分离:人外周血400×g离心15分钟取血沉棕黄层,铺于Ficoll-Paque密度梯度液(1.077 g/mL)上,800×g离心20分钟,收集界面层PBMCs,用RPMI 1640洗涤后重悬至1×10⁶ cells/mL。 2. 处理:PBMCs以1 mL/孔接种24孔板,GSK256066(0.1~10 nM)预处理1小时,再用LPS(1 μg/mL)刺激24小时。 3. 细胞因子检测:收集培养上清,夹心ELISA测TNF-α/IL-6水平 [1] - HBEC细胞MUC5AC表达实验[1]: 1. 细胞培养:HBECs以2×10⁵个/孔接种6孔板,用支气管上皮生长培养基(BEGM)培养至80%融合。 2. 处理:GSK256066(0.05~5 nM)预处理1小时,再用TNF-α(10 ng/mL)刺激16小时。 3. mRNA检测:TRIzol试剂提取总RNA,逆转录为cDNA,实时定量RT-PCR测MUC5AC mRNA(以GAPDH为内参) [1] |
| 动物实验 |
动物/疾病模型:雄性褐挪威大鼠(180-200克)[2]
剂量:10微克/千克 给药途径:管内注射; LPS 刺激前(36 小时、24 小时、18 小时、12 小时、6 小时和 2 小时)和刺激后(0 小时、2 小时) 实验结果:抑制 LPS 诱导的肺中性粒细胞增多。 豚鼠气道舒张试验(参考文献 2): 1. 动物准备:雄性 Dunkin-Hartley 豚鼠(300–350 克,每组 n=6)用氯胺酮(80 毫克/千克,腹腔注射)+ 赛拉嗪(10 毫克/千克,腹腔注射)麻醉,并插入气管插管进行通气。 2. 药物配制:将 GSK256066 溶解于含 0.01% Tween 80 的生理盐水中,配制成浓度分别为 0.01 mg/mL (0.1 mg/kg)、0.03 mg/mL (0.3 mg/kg) 和 0.1 mg/mL (1 mg/kg) 的溶液(基于 10 mL/kg 的雾化体积)。3. 吸入给药:使用连接至气管插管的小容量雾化器(输出速率 0.5 mL/min,粒径 1–5 μm)进行雾化,持续 10 分钟。 4. 气道阻力测量:给药后30分钟,静脉注射组胺(1 mg/kg),并通过连接气管插管的压力传感器,每隔5分钟测量一次气道阻力(Raw),持续30分钟[2] - 大鼠气道炎症试验(文献2): 1. 炎症诱导:雄性Sprague-Dawley大鼠(250-300g,每组n=8)用异氟烷麻醉,并通过22G导管气管内滴注LPS(0.5 mg/kg,溶于0.2 mL生理盐水)。 2. 给药:按上述方法制备GSK256066(0.3 mg/kg,1 mg/kg),并雾化吸入10分钟,每日一次,连续3天(从LPS给药后24小时开始)。 3. 支气管肺泡灌洗液 (BALF) 采集:第 4 天,对大鼠进行颈椎脱臼处死。插入气管,注入 3×1 mL 生理盐水(400×g 离心 10 分钟以分离细胞/细胞因子)采集 BALF [2] |
| 药代性质 (ADME/PK) |
大鼠吸入药代动力学(文献2):
雄性Sprague-Dawley大鼠(每时间点n=4)吸入GSK256066 1 mg/kg(雾化浓度为0.1 mg/mL溶液)。分别于给药后0.25、0.5、1、2、4、8小时采集血浆和肺组织样本: -肺组织浓度:0.25小时达到峰值(1200 ± 150 ng/g),8小时降至150 ± 20 ng/g; -血浆浓度:0.5小时达到峰值(8 ± 1 ng/mL),4小时降至<1 ng/mL; - 肺血浆浓度比:150:1(0.25 小时),300:1(2 小时)[2] - 代谢稳定性(文献 1): 在人/大鼠肝微粒体中,GSK256066 显示出较低的固有清除率 (CLint):2.1 ± 0.3 μL/min/mg 蛋白(人),3.5 ± 0.5 μL/min/mg 蛋白(大鼠)。孵育 60 分钟后,母体药物代谢率低于 10% [1] - 口服生物利用度(文献 1): 大鼠 (n=4) 口服 GSK256066 10 mg/kg(悬浮于 0.5% CMC-Na 中)。血浆AUC0–8h为12±2 ng·h/mL,而静脉注射1 mg/kg时为850±50 ng·h/mL,导致口服生物利用度(F)<2%[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
体外细胞毒性(文献1):
用GSK256066(0.01–100 nM)处理48小时后,HBEC、PBMC和人肝细胞的存活率>90%(MTT法),未见明显的细胞毒性[1] -体内急性毒性(文献2): 大鼠(每组n=4)每日一次吸入GSK256066 5 mg/kg(5倍治疗剂量),连续7天。未观察到死亡、体重减轻(<3%)或异常行为(例如嗜睡、呼吸困难)。血清ALT/AST/BUN/肌酐水平正常;肺组织病理学检查未见炎症或纤维化[2] - 血浆蛋白结合率(文献1): 在人/大鼠血浆中,GSK256066 (0.1–10 nM) 具有较高的蛋白结合率:98.5 ± 0.5%(人),97.8 ± 0.8%(大鼠)(通过超滤法测定)[1] |
| 参考文献 |
|
| 其他信息 |
GSK256066 已用于研究 SAR、哮喘、轻度哮喘、过敏性鼻炎和季节性过敏性鼻炎等疾病的治疗和诊断的试验中。
作用机制: GSK256066 与 PDE4 的催化位点竞争性结合,抑制 cAMP 水解并增加细胞内 cAMP 水平。 cAMP 水平升高可激活蛋白激酶 A (PKA),从而抑制 NF-κB 和 AP-1 信号通路的激活,进而减少促炎细胞因子(TNF-α、IL-6)和粘液相关基因(MUC5AC)的产生 [1,2] - 治疗潜力: GSK256066 是一种吸入式 PDE4 抑制剂,具有高肺部靶向性和低全身暴露量,是治疗呼吸系统炎症性疾病(如哮喘和慢性阻塞性肺疾病 (COPD))的候选药物 [1,2] - 开发优势: 1. 具有卓越的 PDE4 亲和力 (Ki = 0.015 nM) 和亚型选择性(与其他 PDE 相比),最大限度地减少脱靶效应(例如,肠道中 PDE4B 抑制引起的恶心); 2. 吸入给药可使肺部药物浓度达到较高水平(1200 ng/g),而血浆药物浓度较低(<10 ng/mL),从而降低全身毒性;3. 代谢稳定性高(CLint <4 μL/min/mg),支持每日一次给药[1,2] |
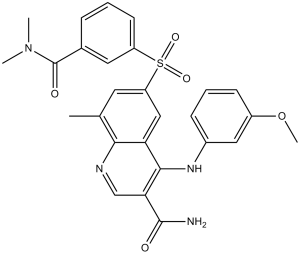
| 分子式 |
C27H26N4O5S
|
|---|---|
| 分子量 |
518.58
|
| 精确质量 |
518.162
|
| CAS号 |
801312-28-7
|
| 相关CAS号 |
GSK256066 Trifluoroacetate;1415560-64-3
|
| PubChem CID |
9827968
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
791.7±60.0 °C at 760 mmHg
|
| 闪点 |
432.6±32.9 °C
|
| 蒸汽压 |
0.0±2.8 mmHg at 25°C
|
| 折射率 |
1.654
|
| LogP |
3.63
|
| tPSA |
144.29
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
37
|
| 分子复杂度/Complexity |
922
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CC1=CC(=CC2=C(C(=CN=C12)C(=O)N)NC3=CC(=CC=C3)OC)S(=O)(=O)C4=CC=CC(=C4)C(=O)N(C)C
|
| InChi Key |
JFHROPTYMMSOLG-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C27H26N4O5S/c1-16-11-21(37(34,35)20-10-5-7-17(12-20)27(33)31(2)3)14-22-24(16)29-15-23(26(28)32)25(22)30-18-8-6-9-19(13-18)36-4/h5-15H,1-4H3,(H2,28,32)(H,29,30)
|
| 化学名 |
6-[[3-[(Dimethylamino)carbonyl]phenyl]sulfonyl]-4-[(3-methoxyphenyl)amino]-8-methyl-3-quinolinecarboxamide
|
| 别名 |
GSK-256066; GSK 256066; GSK256066;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9283 mL | 9.6417 mL | 19.2834 mL | |
| 5 mM | 0.3857 mL | 1.9283 mL | 3.8567 mL | |
| 10 mM | 0.1928 mL | 0.9642 mL | 1.9283 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT00464568
Conditions:Rhinitis, Allergic, SeasonalLink: https://clinicaltrials.gov/ct2/show/NCT00515268
Conditions:Pulmonary Disease, Chronic ObstructiveLink: https://clinicaltrials.gov/ct2/show/NCT00549679
Conditions:Pulmonary Disease, Chronic Obstructive
Title:Clinical Endpoint Trial Investigating Once Daily and Bronchodilator Dosing
Status:Completed
updateDate:2017-06-26
Ctid:NCT00549744
Link: https://clinicaltrials.gov/ct2/show/NCT00549744
Conditions:AsthmaLink: https://clinicaltrials.gov/ct2/show/NCT00612820
Conditions:Rhinitis, Allergic, SeasonalLink: https://clinicaltrials.gov/ct2/show/NCT00377728
Conditions:Rhinitis, Allergic, Seasonal|Seasonal Allergic RhinitisLink: https://clinicaltrials.gov/ct2/show/NCT00612118
Conditions:Allergic Rhinitis|Rhinitis, Allergic, SeasonalLink: https://clinicaltrials.gov/ct2/show/NCT00380354
Conditions:Asthma|Mild AsthmaLink: https://clinicaltrials.gov/ct2/show/NCT00445510
Conditions:AsthmaLink: https://clinicaltrials.gov/ct2/show/NCT00430157
Conditions:Rhinitis, Allergic, Seasonal|Seasonal Allergic Rhinitis
|
|---|
|
|
|
|---|
|
|
 FCPR-16
FCPR-16
 Mardepodect盐酸盐
Mardepodect盐酸盐
 盐酸阿那格雷
盐酸阿那格雷
 己酮可可碱
己酮可可碱
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA






 463611831
463611831
