| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 2mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
Akt1 (IC50 = 5 nM); Akt3 (IC50 = 8 nM); Akt2 (IC50 = 18 nM); PKA (IC50 = 3100 nM)
Selective ATP-competitive inhibitor of Akt isoforms, including Ipatasertib (GDC0068; RG7440). It exhibits potent inhibitory activity against Akt1 with an IC50 of approximately 5 nM, Akt2 with an IC50 of approximately 18 nM, and Akt3 with an IC50 of approximately 12 nM. It shows high selectivity over other kinases (e.g., >100-fold selectivity against PKA, PKC, and other AGC kinases) [1] - Ipatasertib (GDC0068; RG7440) maintains selective inhibition of Akt1 (IC50: 5 nM), Akt2 (IC50: 18 nM), and Akt3 (IC50: 12 nM) in kinase panels, with minimal activity against non-Akt kinases, confirming its specificity for the Akt family [2] - In cellular assays, Ipatasertib (GDC0068; RG7440) inhibits phosphorylated Akt (p-Akt) at Ser473 and Thr308, downstream of Akt isoforms, without affecting the total Akt protein level; no new IC50 values for Akt isoforms are provided beyond those reported in [1] [3] |
|---|---|
| 体外研究 (In Vitro) |
当针对大量 230 种激酶(PRKG1α、PRKG1β 和 p70S6K,IC50 值分别为 98 nM、69 nM 和 860 nM)进行测试时,GDC-0068 在 1 μM 浓度下仅抑制 3 种激酶 >70%。 GDC-0068 的 IC50 为 3.1 μM,对 Akt 的选择性是 PKA 的 100 倍以上。 GDC-0068 处理可抑制 LNCaP、PC3 和 BT474M1 细胞中 Akt 底物 PRAS40 的磷酸化,IC50 值分别为 157 nM、197 nM 和 208 nM。此外,GDC-0068 选择性抑制肿瘤抑制因子 PTEN 缺陷、PIK3CA 致癌突变和 HER2 扩增的 Akt 信号驱动癌细胞系,其中 HER2+ 和 Luminal 亚型的效果最强。 [1-3]
Ipatasertib (GDC0068; RG7440) 对多种Akt信号激活的人肿瘤细胞系具有增殖抑制作用。例如,对PC-3前列腺癌细胞的抗增殖IC50约为0.8 μM;对LNCaP前列腺癌细胞的IC50约为1.2 μM;对MDA-MB-468乳腺癌细胞的IC50约为0.6 μM。Western blot结果显示,用1-5 μM Ipatasertib (GDC0068; RG7440) 处理细胞4小时,可使p-Akt(Ser473和Thr308)水平降低>70%,同时使下游底物(如p-FOXO1 Ser256、p-GSK3β Ser9)的磷酸化水平降低60%-80%[1] - 在24种人肿瘤细胞系组成的筛选面板中,Ipatasertib (GDC0068; RG7440) 的抗增殖IC50范围为0.3 μM-3.5 μM,其中对PTEN缺失或PI3K突变的细胞系(如BT-474乳腺癌细胞)活性最强,IC50为0.3 μM。Ipatasertib (GDC0068; RG7440)(0.5 μM)与紫杉醇(10 nM)联合处理MDA-MB-231乳腺癌细胞,较单药处理细胞活力额外降低40%。克隆形成实验显示,0.5 μM Ipatasertib (GDC0068; RG7440) 处理PC-3细胞14天后,克隆形成率降低>60%[2] - Ipatasertib (GDC0068; RG7440) 可诱导HCT116结直肠癌细胞和MCF-7乳腺癌细胞凋亡。2 μM浓度处理24小时后,凋亡细胞比例(Annexin V阳性)较对照组增加30%-40%。Western blot显示,1-3 μM Ipatasertib (GDC0068; RG7440) 可使促凋亡蛋白PUMA表达上调2-3倍,并激活Caspase-3/7(切割型Caspase-3水平增加>2倍)。实时定量PCR结果显示,2 μM Ipatasertib (GDC0068; RG7440) 处理HCT116细胞后,PUMA mRNA表达上调3.5倍。此外,敲低FoxO3a或NF-κB p65可消除Ipatasertib (GDC0068; RG7440) 诱导的PUMA上调,表明FoxO3a和NF-κB介导该效应[3] |
| 体内研究 (In Vivo) |
GDC-0068 口服给药会导致 PC3 前列腺肿瘤异种移植模型中 p-PRAS40 下调。 BT474-Tr 异种移植物中的 GDC-0068 治疗可降低 pS6 和 peIF4G 水平,将 FOXO3a 重新定位到细胞核,并导致 HER3 和 pERK 的反馈上调。在许多异种移植肿瘤模型中,包括 PTEN 缺陷型前列腺癌模型 LNCaP 和 PC3、PIK3CA H1047R 突变乳腺癌模型 KPL-4 和 MCF7-neo/HER2 肿瘤模型,GDC-0068 的给药表现出强大的抗肿瘤功效。 [1-3]
在PC-3前列腺癌裸鼠异种移植模型中,口服给予Ipatasertib (GDC0068; RG7440)(30 mg/kg/天、100 mg/kg/天)21天,肿瘤生长抑制率(TGI)分别为45%和72%(vs 溶媒对照组)。处理组小鼠无明显体重下降(≤5%)。肿瘤组织Western blot显示,100 mg/kg/天Ipatasertib (GDC0068; RG7440) 可使p-Akt(Ser473)水平降低>80%,p-GSK3β(Ser9)水平降低70%[1] - 在MDA-MB-468乳腺癌裸鼠异种移植模型中,Ipatasertib (GDC0068; RG7440) 单药(100 mg/kg/天,口服)处理28天,TGI为65%;与多西他赛(10 mg/kg/周,腹腔注射)联合处理,TGI达90%,且显著延长小鼠中位生存期(溶媒组42天 vs 联合组68天)。肿瘤组织免疫组化(IHC)显示,Ipatasertib (GDC0068; RG7440) 处理使p-Akt(Ser473)染色强度降低>70%[2] - 在HCT116结直肠癌裸鼠异种移植模型中,口服Ipatasertib (GDC0068; RG7440)(100 mg/kg/天)14天,肿瘤组织中PUMA蛋白表达(IHC检测)较对照组增加2.5倍,凋亡细胞数(TUNEL阳性)增加3倍。处理组小鼠无明显毒性(如体重下降、器官损伤)[3] |
| 酶活实验 |
Enzymatic Assays/酶实验[1]
用于测定Akt1/2/3和PKA激酶活性的测定采用IMAP荧光偏振(FP)磷酸化检测试剂来检测已被相应激酶磷酸化的荧光标记肽底物。这些研究中使用的Akt酶由重组杆状病毒表达、氨基末端、聚组氨酸标记、全长、野生型人类形式组成,并从商业上获得。这些研究中使用的PKA酶由商业上获得的大肠杆菌中表达的重组未标记的PKA人分离催化亚基组成。在环境温度下,将抑制剂、酶(9 nM Akt1或100 pM PKA)和底物(100 nM Crosstide)与5μM ATP在测定缓冲液(10 mM Tris-HCl(pH 7.2)、10 mM MgCl2、0.1%BSA(w/v)、最终DMSO 2%(v/v))中在5μL反应体积中孵育60分钟。通过向ATP溶液中加入酶+肽底物引发反应。加入IMAP结合试剂(15μL)终止反应,并将停止的反应在室温下孵育至少30分钟。 Akt激酶活性实验(基于HTRF技术):将重组人Akt亚型(Akt1、Akt2、Akt3)用实验缓冲液(含Tris-HCl、MgCl2、DTT和BSA)稀释至终浓度1 nM;向反应体系中加入底物(生物素化GSK3β肽段,终浓度2 μM)、ATP(终浓度10 μM,接近Akt的Km值)及不同浓度的Ipatasertib (GDC0068; RG7440)(0.1 nM-10 μM);30°C孵育60分钟后,加入检测混合物(链霉亲和素偶联Eu3+与抗磷酸化GSK3β抗体偶联XL665);检测665 nm和620 nm处的荧光共振能量转移(FRET)信号,以665/620比值计算激酶活性抑制率;采用四参数逻辑模型拟合抑制曲线,计算IC50[1] - 激酶选择性实验:使用包含130种重组人激酶(含AGC家族激酶、酪氨酸激酶、丝氨酸/苏氨酸激酶)的筛选面板;每种激酶与特异性底物、ATP(Km浓度)及1 μM Ipatasertib (GDC0068; RG7440) 共同孵育;采用放射性法(33P-ATP掺入底物)检测激酶活性;以溶媒对照组为参照,计算剩余激酶活性百分比,活性剩余<10%的激酶视为潜在脱靶;结果显示仅Akt1、Akt2、Akt3活性剩余<10%,证实Ipatasertib (GDC0068; RG7440) 的选择性[1] - 细胞裂解液中p-Akt抑制实验:用Ipatasertib (GDC0068; RG7440) 处理肿瘤细胞(如PC-3)4小时后,用含蛋白酶和磷酸酶抑制剂的RIPA缓冲液裂解细胞;离心收集上清液,取20 μg蛋白进行SDS-PAGE电泳,转印至PVDF膜;用5%脱脂牛奶封闭膜后,4°C孵育抗p-Akt(Ser473)、p-Akt(Thr308)或总Akt一抗过夜;洗涤后,室温孵育HRP偶联二抗1小时;ECL底物显影,ImageJ软件定量条带强度;以溶媒对照组为参照,计算p-Akt抑制率[2] |
| 细胞实验 |
在以5000个细胞/孔的密度镀在黑色透明底96孔板上的LNCaP细胞中测量细胞存活率的抑制,随后在37°C和5%CO2下用0-10μM 28(ipatasertib)处理72小时。细胞增殖的程度是通过使用560nm的激发波长和590nm的发射波长测量制造商方案中所述的刃天青还原为再硫酸镁来确定的。使用四参数逻辑斯蒂模型生成剂量-反应曲线,并根据这些曲线拟合确定50%抑制浓度(IC50)值。[1]
简而言之,在ipatasertib处理后,HCT116 WT或p53-/-用1%甲醛固定,并在温SDS裂解缓冲液中裂解。获得基因组DNA,并通过在冰上超声处理将其剪切至200-1000bp。样品用蛋白A-琼脂糖/鲑鱼精子DNA(50%浆液)在4°C下搅拌预清洗1小时。然后加入抗FoxO3a抗体或抗p65抗体,在4°C的摇床上孵育过夜。正常兔IgG用作阴性对照。然后加入蛋白质琼脂糖/鲑鱼精子DNA(50%浆液)珠,以沉淀抗体/蛋白质/DNA复合物。用连续洗涤缓冲液洗涤后,用洗脱缓冲液(1%SDS,0.1 M NaHCO3)从珠粒中洗脱DNA-蛋白质免疫复合物30分钟。最后,通过在65°C下与0.2 M NaCl孵育4小时,逆转蛋白质-DNA交联以释放DNA[3]。 抗增殖实验(MTT法):将人肿瘤细胞以2×10^3个/孔的密度接种于96孔板,过夜培养;向各孔加入不同浓度的Ipatasertib (GDC0068; RG7440)(0.01 μM-10 μM),每个浓度设3个复孔;37°C、5% CO2培养箱中孵育72小时;每孔加入20 μL MTT溶液(5 mg/mL),继续孵育4小时;弃去上清液,加入150 μL DMSO溶解甲瓒结晶;酶标仪检测570 nm处吸光度;以溶媒对照组为参照计算细胞活力,GraphPad Prism软件计算IC50[1] - 克隆形成实验:PC-3细胞以5×10^2个/孔的密度接种于6孔板,过夜培养;加入终浓度为0.1 μM、0.5 μM、1 μM的Ipatasertib (GDC0068; RG7440),设溶媒对照组;37°C、5% CO2培养箱中培养14天,每3天更换培养基和药物;培养结束后,4%多聚甲醛固定细胞15分钟,0.1%结晶紫染色30分钟,水洗;显微镜下计数含>50个细胞的克隆;克隆形成率=(药物组克隆数/对照组克隆数)×100%[2] - 凋亡实验(Annexin V-FITC/PI双染法):HCT116细胞以2×10^5个/孔接种于6孔板,过夜培养;加入终浓度2 μM的Ipatasertib (GDC0068; RG7440),孵育24小时;胰酶消化收集细胞,冷PBS洗涤2次,用1×结合缓冲液重悬;向细胞悬液中加入5 μL Annexin V-FITC和10 μL PI,室温避光孵育15分钟;流式细胞仪检测凋亡率,FlowJo软件分析数据[3] - PUMA mRNA实时定量PCR检测:2 μM Ipatasertib (GDC0068; RG7440) 处理HCT116细胞12小时;RNA提取试剂盒提取总RNA,NanoDrop分光光度计测定RNA浓度和纯度;逆转录试剂盒合成cDNA;使用SYBR Green PCR预混液进行实时定量PCR,以PUMA特异性引物和GAPDH特异性引物(内参)扩增;反应条件:95°C 10分钟,随后40个循环(95°C 15秒、60°C 1分钟);采用2^(-ΔΔCt)法计算PUMA mRNA相对表达量[3] |
| 动物实验 |
携带 LNCaP、PC3、KPL-4 或 MCF7 肿瘤异种移植的雌性裸鼠
~100 mg/kg/天 口服 体内肿瘤异种移植研究中,将悬浮于 Hank's 平衡盐溶液 (HBSS) 中的 PC3 细胞皮下接种于雌性裸鼠 (nu/nu) 的右后侧。当肿瘤平均体积达到 150 mm³ 时,将动物按大小进行匹配,并随机分配到各治疗组,每组 10 只动物。肿瘤体积计算如下:肿瘤大小 (mm³) = (较长测量值 × (较短测量值)²) × 0.5。数据分析后,使用 JMP 统计软件 7.0 版 (SAS Institute) 进行 Dunnett's t 检验,确定 p 值。使用 Adventura Pro AV812 电子秤 (Ohaus Corp.) 每周两次记录小鼠体重。根据IACUC方案指南[1],当肿瘤体积超过2000 mm³或体重下降≥初始体重的20%时,立即对小鼠实施安乐死。 在PK/PD研究中,从荷瘤PC3小鼠单次注射ipatasertib后1、3、8和24小时采集血液和肿瘤样本。在预定的样本采集时间,通过心脏穿刺从每只动物采集约800 μL血液样本,置于含有K2EDTA抗凝剂的试管中,并在1500–2000 g下离心以分离血浆。使用未经验证的LC/MS/MS方法,在基因泰克公司DMPK生物分析部门测定每个血浆样本中ipatasertib的浓度。该方法的定量下限(LLOQ)为0.005 μM。将肿瘤样本在含有 150 mM NaCl、20 mM Tris (pH 7.5)、1 mM EDTA、1 mM EGTA 和 1% Triton X-100 的 Tris 裂解缓冲液中进行解离。使用 BCA 蛋白测定试剂盒测定蛋白浓度。采用人酶联免疫吸附试验 (ELISA) 试剂盒测定总 PRAS40 和 Thr246 位点磷酸化 PRAS40 (p-PRAS40) 的水平。该检测方法基于吸光度测量来定量蛋白水平。显色产物的强度与样本中 p-PRAS40 和 tPRAS40 的浓度成正比。使用 Meso Scale Discovery 多点生物标志物检测系统测定总 S6RP 和 Ser235/236 位点磷酸化 S6RP (pS6RP) 的水平。这些检测方法基于电化学发光强度测量来定量蛋白水平。在接受ipatasertib治疗的肿瘤中,磷酸化蛋白水平以总蛋白水平进行标准化,并与载体对照组进行比较。[1] 在多种肿瘤细胞系和患者来源的异种移植模型中评估了体内疗效。将细胞或肿瘤碎片皮下植入免疫缺陷小鼠的侧腹部。使用雌性或雄性裸鼠(nu/nu)或严重联合免疫缺陷小鼠(SCID)/米色小鼠。对于MCF7-neo/HER2模型,在细胞接种前,将17β-雌二醇缓释片(0.36 mg/片,60天释放,货号SE-121)植入小鼠背肩部。雄性小鼠在植入肿瘤碎片前进行去势。将肿瘤细胞或碎片植入小鼠体内后,监测肿瘤直至平均肿瘤体积达到 180 至 350 mm³,然后将其分为 8 至 10 只/组。ipatasertib/GDC-0068 配制于 0.5% 甲基纤维素/0.2% Tween-80 (MCT) 溶液中,每日一次 (QD) 通过灌胃法给药。[2] 收集 HCT116 WT 和 PUMA−/− 细胞,将 1 × 10⁶ 个细胞悬浮于 0.2 ml 培养基中,皮下注射到无胸腺裸鼠背部。5 周龄雌性裸鼠饲养于无菌微隔离笼中,可自由摄取水和饲料。小鼠在接受为期7天的治疗后,每日灌胃给予40 mg/kg的ipatasertib/GDC-0068,持续21天。使用游标卡尺监测肿瘤生长情况,肿瘤体积按公式0.5 × 长 × 宽²计算。当肿瘤体积达到约1.0 cm³时,对小鼠实施安乐死。解剖肿瘤,用10%福尔马林固定,并进行石蜡包埋。[3] 裸鼠PC-3前列腺癌异种移植模型:使用6-8周龄的雄性裸鼠。将处于对数生长期的PC-3细胞(1×10^6个细胞,溶于0.1 mL PBS/Matrigel混合液,1:1)皮下注射到每只小鼠的右侧背部。当肿瘤体积达到 100-200 mm³ 时,将小鼠随机分为 3 组(每组 n=6):载体对照组、Ipatasertib (GDC0068; RG7440) 30 mg/kg 组和 Ipatasertib (GDC0068; RG7440) 100 mg/kg 组。Ipatasertib (GDC0068; RG7440) 溶于由 0.5% 甲基纤维素和 0.1% Tween 80 组成的载体中,每日口服一次。载体对照组给予等体积的载体。每周测量两次肿瘤体积(计算公式为长 × 宽² × 0.5)和小鼠体重。治疗21天后,处死小鼠,收集肿瘤组织进行Western blot分析[1] - MDA-MB-468乳腺癌异种移植联合模型:将MDA-MB-468细胞(2×10^6个细胞,溶于0.1 mL PBS/Matrigel,1:1)皮下注射到6-8周龄雌性裸鼠的右侧背部。当肿瘤体积达到150-200 mm³时,将小鼠分为4组(每组n=8):载体对照组、单独使用Ipatasertib(GDC0068;RG7440)(100 mg/kg,口服,每日一次)、单独使用多西他赛(10 mg/kg,腹腔注射,每周一次)以及Ipatasertib(GDC0068;RG7440)联合多西他赛治疗组。治疗持续28天。每周测量两次肿瘤体积和体重。每日记录小鼠存活情况,并计算中位生存时间。治疗后,收集肿瘤组织进行p-Akt的免疫组织化学(IHC)分析[2] - HCT116结直肠癌异种移植机制模型:将HCT116细胞(1.5×10^6个细胞溶于0.1 mL PBS/Matrigel,1:1)皮下注射到6-8周龄的裸鼠体内。当肿瘤体积达到100-150 mm³时,将小鼠分为两组(每组n=6):载体对照组和Ipatasertib(GDC0068;RG7440)100 mg/kg(口服,每日一次)组。治疗持续14天。每3天测量一次肿瘤体积和体重。处死小鼠后,将肿瘤组织固定于4%多聚甲醛溶液中,石蜡包埋,并进行切片。采用免疫组化法(IHC)检测PUMA蛋白表达(使用抗PUMA抗体),并采用TUNEL染色检测凋亡细胞。在显微镜下计数PUMA阳性细胞和TUNEL阳性细胞的数量[3] |
| 药代性质 (ADME/PK) |
体外代谢:将人肝微粒体(0.5 mg/mL)与伊帕他替尼(GDC0068;RG7440)(1 μM)和 NADPH(1 mM)在 37°C 下孵育 0-60 分钟。加入含有内标的乙腈终止反应。离心后,取上清液进行 LC-MS/MS 分析,以检测代谢产物。结果表明,伊帕他替尼(GDC0068;RG7440)主要通过 CYP3A4 代谢,产生三种主要的氧化代谢产物(M1、M2 和 M3)。伊帕他替尼(GDC0068;RG7440)在人肝微粒体中的半衰期约为45分钟[1]
- 血浆蛋白结合率:将伊帕他替尼(GDC0068;RG7440)(1 μM)与人、大鼠和犬血浆(0.5 mL)在37°C下孵育1小时。然后使用离心超滤管对样品进行超滤。采用液相色谱-串联质谱法(LC-MS/MS)测定滤液(游离药物)和血浆(总药物)中伊帕他替尼(GDC0068;RG7440)的浓度。血浆蛋白结合率计算公式为:[(总药物浓度 - 游离药物浓度) / 总药物浓度] × 100%。结果显示,在人、大鼠和犬血浆中,血浆蛋白结合率均>95%[1] - 大鼠体内药代动力学:雄性Sprague-Dawley大鼠(每个时间点n=3)单次口服伊帕他替尼(GDC0068;RG7440)(10 mg/kg,溶于0.5%甲基纤维素/0.1%吐温80)或静脉注射(2 mg/kg,溶于DMSO/生理盐水,1:9)。分别于给药后0.25、0.5、1、2、4、6、8、12和24小时采集血样。通过离心分离血浆,并采用液相色谱-串联质谱法(LC-MS/MS)测定伊帕他替尼(GDC0068;RG7440)的浓度。采用非房室模型分析计算药代动力学参数:口服生物利用度 (F) 为 35%,Tmax(口服)为 1.5 小时,Cmax(口服)为 1.2 μg/mL,末端半衰期 (t1/2) 为 5.2 小时 [1] - 小鼠体内药代动力学:雌性裸鼠(每个时间点 n=3)单次口服伊帕他替尼 (GDC0068; RG7440) (100 mg/kg)。分别于 0.25、0.5、1、2、4、6、8 和 12 小时采集血样。血浆浓度分析显示 Tmax 为 1 小时,Cmax 为 5.1 μg/mL,t1/2 为 3.1 小时 [2] |
| 毒性/毒理 (Toxicokinetics/TK) |
小鼠急性毒性试验:雌性ICR小鼠(每组n=5)单次口服500、1000或2000 mg/kg的Ipatasertib(GDC0068;RG7440)。观察小鼠14天,记录死亡率、临床症状和体重变化。各组均未观察到死亡。在1000和2000 mg/kg剂量组中,前3天内观察到轻微且短暂的体重下降(≤5%),但体重在第7天恢复正常。未观察到其他毒性临床症状(例如,嗜睡、腹泻)[1]。
- 大鼠重复给药毒性:雄性Sprague-Dawley大鼠(每组n=6)口服Ipatasertib(GDC0068;RG7440),剂量分别为10、30或100 mg/kg/天,持续28天。赋形剂对照组给予0.5%甲基纤维素/0.1%吐温80。每周测量体重,并在治疗结束时采集血样进行临床化学分析。在 100 mg/kg/天的剂量下,观察到血清 ALT(较对照组升高 1.5 倍)和 AST(较对照组升高 1.3 倍)略有升高,但未检测到肝脏组织病理学改变。在 10 mg/kg/天和 30 mg/kg/天的剂量组中,未观察到体重、血液学参数或其他临床化学指标的显著变化。未发生与毒性相关的死亡[1] - 犬的毒代动力学:比格犬(每组 n=4)分别口服 5、20 或 50 mg/kg/天的伊帕他替尼(GDC0068;RG7440),持续 90 天。分别于第 1 天和第 90 天采集血浆样本,以测定伊帕他替尼(GDC0068;RG7440)的浓度。 Cmax 和 AUC0-24h 随剂量增加呈比例增加,未见蓄积(蓄积比 <1.2)。各组均未观察到体重、临床症状或实验室参数的显著变化,表明在高达 50 mg/kg/天的剂量下具有良好的耐受性 [2] |
| 参考文献 |
|
| 其他信息 |
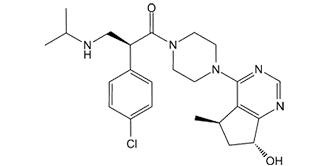
(2S)-2-(4-氯苯基)-1-[4-[(5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊并[d]嘧啶-4-基]-1-哌嗪基]-3-(丙-2-基氨基)-1-丙酮是一种N-芳基哌嗪类化合物。
伊帕他替尼已用于多种癌症、肿瘤、实体瘤、乳腺癌和胃癌等的治疗试验。 伊帕他替尼是一种口服生物利用度高的丝氨酸/苏氨酸蛋白激酶Akt(蛋白激酶B)抑制剂,具有潜在的抗肿瘤活性。伊帕他替尼以非ATP竞争性方式结合并抑制Akt的活性,这可能导致PI3K/Akt信号通路和肿瘤细胞增殖的抑制,并诱导肿瘤细胞凋亡。 PI3K/Akt信号通路的激活通常与肿瘤发生相关,而PI3K/Akt信号通路的失调可能导致肿瘤对多种抗肿瘤药物产生耐药性。 药物适应症 治疗乳腺癌,治疗前列腺癌 Ipatasertib (GDC0068; RG7440) 被开发为一种靶向治疗药物,用于治疗Akt异常激活的肿瘤,这种异常激活通常由PTEN缺失、PI3K突变或Akt扩增等基因改变引起。这些改变导致细胞不受控制地增殖和存活,使 Akt 成为肿瘤学中的关键治疗靶点 [1] - 在临床前研究中,Ipatasertib (GDC0068; RG7440) 对多种 Akt 激活的肿瘤类型显示出疗效,包括前列腺癌、乳腺癌、结直肠癌和肺癌,这支持了其在癌症治疗中广泛应用的潜力 [2] - Ipatasertib (GDC0068; RG7440) 诱导细胞凋亡的机制涉及对 PUMA 的双重调控:抑制 Akt 可使 FoxO3a 从细胞质隔离中释放出来,使其易位到细胞核并反式激活 PUMA;同时,Ipatasertib (GDC0068; RG7440) 激活 NF-κB,NF-κB 直接与 PUMA 启动子结合并增强 PUMA 转录。这种双重机制促成了伊帕他替尼(GDC0068;RG7440)对肿瘤细胞的强效凋亡作用[3] |
| 分子式 |
C24H32CLN5O2
|
|---|---|
| 分子量 |
457.9962
|
| 精确质量 |
457.224
|
| 元素分析 |
C, 62.94; H, 7.04; Cl, 7.74; N, 15.29; O, 6.99
|
| CAS号 |
1001264-89-6
|
| 相关CAS号 |
Ipatasertib dihydrochloride;1396257-94-5; Ipatasertib;1001264-89-6; 1489263-16-2 (HCl); 1491138-24-9; 1491138-23-8 (besylate)
|
| PubChem CID |
24788740
|
| 外观&性状 |
White to light yellow solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
669.4±55.0 °C at 760 mmHg
|
| 闪点 |
358.7±31.5 °C
|
| 蒸汽压 |
0.0±2.1 mmHg at 25°C
|
| 折射率 |
1.603
|
| LogP |
1.71
|
| tPSA |
81.59
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
32
|
| 分子复杂度/Complexity |
622
|
| 定义原子立体中心数目 |
3
|
| SMILES |
ClC1C([H])=C([H])C(=C([H])C=1[H])[C@@]([H])(C([H])([H])N([H])C([H])(C([H])([H])[H])C([H])([H])[H])C(N1C([H])([H])C([H])([H])N(C([H])([H])C1([H])[H])C1C2=C([C@@]([H])(C([H])([H])[C@@]2([H])C([H])([H])[H])O[H])N=C([H])N=1)=O
|
| InChi Key |
GRZXWCHAXNAUHY-NSISKUIASA-N
|
| InChi Code |
InChI=1S/C24H32ClN5O2/c1-15(2)26-13-19(17-4-6-18(25)7-5-17)24(32)30-10-8-29(9-11-30)23-21-16(3)12-20(31)22(21)27-14-28-23/h4-7,14-16,19-20,26,31H,8-13H2,1-3H3/t16-,19-,20-/m1/s1
|
| 化学名 |
(2S)-2-(4-chlorophenyl)-1-[4-[(5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl]piperazin-1-yl]-3-(propan-2-ylamino)propan-1-one
|
| 别名 |
GDC0068; GDC 0068; GDC-0068; RG-7440; 1001264-89-6; Ipatasertib (GDC-0068); RG7440; (S)-2-(4-chlorophenyl)-1-(4-((5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl)piperazin-1-yl)-3-(isopropylamino)propan-1-one; RG-7440; GDC0068; RG 7440; RG7440; Ipatasertib
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~92 mg/mL (~200.9 mM)
Water: <1 mg/mL Ethanol: ~92 mg/mL (~200.9 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (4.54 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (4.54 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (4.54 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 5% DMSO+40% PEG 300+5%Tween80+ 50%ddH2O: 92mg/ml 配方 5 中的溶解度: 10 mg/mL (21.83 mM) in 0.5% MC 0.5% Tween-80 (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1834 mL | 10.9170 mL | 21.8341 mL | |
| 5 mM | 0.4367 mL | 2.1834 mL | 4.3668 mL | |
| 10 mM | 0.2183 mL | 1.0917 mL | 2.1834 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Targeted Therapy Directed by Genetic Testing in Treating Patients With Advanced Refractory Solid Tumors, Lymphomas, or Multiple Myeloma (The MATCH Screening Trial)
CTID: NCT02465060
Phase: Phase 2 Status: Active, not recruiting
Date: 2024-11-18
Dose-dependent effect of GDC-0068 on Akt pathway biomarkers. Clin Canc Res 2013, 19:1760–1772. |
Single agent efficacy of GDC-0068 in human tumor xenograft models. |
Pharmacokinetic (PK) and pharmacodynamic (PD) relationship of GDC-0068 in xenograft models. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
")
")
")
 463611831
463611831
