| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
20S proteasome (IC50 = 3.4 nM); 20S proteasome (Ki = 0.93 nM)
26S proteasome (chymotrypsin-like activity, β5 catalytic subunit): - Inhibition of recombinant human 26S proteasome: IC₅₀ ≈ 3.4 nM [1] - Selectivity over other proteasome subunits: β1 subunit (caspase-like activity) IC₅₀ ≈ 340 nM, β2 subunit (trypsin-like activity) IC₅₀ ≈ 880 nM, showing >100-fold selectivity for the β5 subunit [1] |
|---|---|
| 体外研究 (In Vitro) |
在较高浓度下,MLN2238 还抑制 caspase 样 (β1) 和胰蛋白酶样 (β2) 蛋白水解位点,IC50 分别为 31nM 和 3.5uM。 MLN2238 抑制 Calu-6 细胞,IC50 为 9.7 nM。 MLN2238 是肿瘤细胞中蛋白酶体的选择性、有效且可逆的抑制剂。 MLN2238 显示出时间依赖性可逆蛋白酶体抑制作用。 MLN2238 和 Bortezomib 均表现出时间依赖性可逆蛋白酶体抑制作用;然而,MLN2238 的蛋白酶体解离半衰期比硼替佐米快 6 倍(分别为 18 分钟和 110 分钟)。 MLN2238 与蛋白酶体的解离速度比硼替佐米更快,这与 Proteasome-Glo 测定中观察到的蛋白酶体活性更快恢复一致。根据 20S 抑制评估,MLN2238 比硼替佐米具有更大的总体肿瘤药效作用。 MLN2238 是 MLN9708 的生物活性形式。激酶测定:Calu-6 细胞在含有 10% 胎牛血清和 1% 青霉素/链霉素的 MEM 中培养,并在实验开始前 1 天以每孔 1 × 104 个细胞接种到 384 孔板中。根据制造商的说明,使用 Proteasome-Glo 测定试剂,通过监测胰凝乳蛋白酶样底物 Suc-LLVY-氨基荧光素在荧光素酶存在下的水解来评估蛋白酶体活性。使用 LEADseeker 仪器测量发光。细胞测定:Calu-6 细胞在含有 10% FBS 和 1% 青霉素/链霉素的 MEM 中培养,并在实验开始前 1 天以每孔 1 × 104 个细胞接种到 384 孔板中。对于 IC50 测定,细胞用不同浓度的 Bortezomib 或 MLN2238 的 DMSO(最终浓度为 0.5%,v/v)在 37 °C 下处理 1 小时。对于可逆性实验,将细胞用 1 μM Bortezomib 或 MLN2238 在 37 °C 下处理 30 分钟,然后在培养基中洗涤三次以去除 Bortezomib 或 MLN2238。将细胞在 37°C 下再孵育 4 小时,然后除去培养基并更换为新鲜培养基。
血液系统恶性肿瘤细胞抗增殖活性(文献[1]、[2]): 1. 多发性骨髓瘤(MM)细胞系(RPMI 8226、MM.1S、U266、OPM-2):IXAZOMIB (MLN2238)(0.1 nM–100 nM,72小时MTT法)浓度依赖性抑制增殖。IC₅₀:RPMI 8226约5 nM,MM.1S约4.2 nM,U266约6.5 nM,OPM-2约5.8 nM。20 nM浓度下,RPMI 8226细胞活力较溶剂对照组降低~75%[1] 2. 硼替佐米耐药MM细胞(RPMI 8226/BtzR):IC₅₀≈8.3 nM(72小时MTT法),对难治性细胞仍保持活性[2] 3. 套细胞淋巴瘤(MCL)细胞系(Granta-519、Jeko-1):IC₅₀分别为~7.1 nM(Granta-519)和~8.5 nM(Jeko-1)(72小时MTT法);15 nM IXAZOMIB (MLN2238)使软琼脂克隆形成率降低~80%[2] - 凋亡诱导(文献[1]、[2]): 1. RPMI 8226细胞:15 nM IXAZOMIB (MLN2238)处理48小时后,凋亡率从对照组的~4%升至~42%(Annexin V-FITC/PI染色,流式细胞术)。Western blot显示活化caspase-3(上调3.2倍)和活化PARP(上调2.8倍)[1] 2. Granta-519细胞:20 nM IXAZOMIB (MLN2238)处理48小时诱导~38%细胞凋亡;TUNEL染色证实DNA片段化(较对照组上调4倍)[2] - NF-κB通路抑制: 1. MM.1S细胞:10 nM IXAZOMIB (MLN2238)(处理24小时)阻断TNF-α诱导的NF-κB激活。Western blot显示,因蛋白酶体降解减少,IκBα蛋白积累(上调4.5倍);免疫荧光染色显示核内p65转位减少~60%[1] - 与来那度胺的协同活性: 1. MM.1S细胞:IXAZOMIB (MLN2238)(5 nM)联合来那度胺(1 μM)使细胞活力降低~85%(单独用药仅降低~30%),联合指数(CI)<1(协同效应)[2] |
| 体内研究 (In Vivo) |
MLN2238 在异种移植肿瘤中诱导比硼替佐米更强的药效学反应。在异种移植模型中,与硼替佐米相比,MLN2238 显示出更大的最大和持续的肿瘤蛋白酶体抑制作用。这些结果证实,MLN2238 改善的肿瘤暴露转化为蛋白酶体水平和下游的肿瘤药效学反应的改善。 MLN2238 在 CWR22 异种移植模型中显示出抗肿瘤活性。与硼替佐米相比,MLN2238 在 WSU-DLCL2 异种移植物中显示出更大的肿瘤药效学反应。同样,硼替佐米治疗仅导致 WSU-DLCL2 异种移植肿瘤中 GADD34 水平轻微增加,而 MLN2238 强烈诱导其表达。在 OCI-Ly10 和 PHTX22L 模型中,与硼替佐米相比,MLN2238 具有改善的药效学特征和抗肿瘤活性
裸鼠MM异种移植模型(文献[1]、[2]): 1. RPMI 8226异种移植: - 分组:小鼠(n=6/组)随机分为3组:(1)对照组(腹腔注射5% DMSO+95%生理盐水);(2)IXAZOMIB (MLN2238) 0.3 mg/kg组;(3)IXAZOMIB (MLN2238) 1 mg/kg组[1] - 给药:肿瘤体积达~100 mm³时开始,腹腔注射给药,每日1次,持续21天[1] - 疗效:肿瘤体积较对照组分别减少~45%(0.3 mg/kg)和~75%(1 mg/kg);肿瘤重量分别降低~40%(0.3 mg/kg)和~70%(1 mg/kg);肿瘤内蛋白酶体β5活性分别抑制~50%(0.3 mg/kg)和~80%[1] 2. MM.1S异种移植+来那度胺联合: - 分组:(1)对照组;(2)IXAZOMIB (MLN2238) 0.5 mg/kg(腹腔注射,每日1次);(3)来那度胺25 mg/kg(口服灌胃,每日1次);(4)联合组[2] - 疗效:第28天联合组肿瘤体积减少~80%(IXAZOMIB (MLN2238)单独组减少~45%,来那度胺单独组减少~40%);血清M蛋白减少~70%(单独组减少~35%)[2] - 小鼠播散性MM模型: 1. 模型构建:向C57BL/KaLwRij小鼠静脉注射5TGM1 MM细胞(1×10⁶个细胞/只)[2] 2. 给药:细胞注射7天后开始,IXAZOMIB (MLN2238) 0.75 mg/kg(腹腔注射,每日1次),持续21天[2] 3. 疗效:骨髓MM细胞浸润(CD138+细胞)减少~60%;骨损伤(micro-CT)较对照组减少~55%[2] |
| 酶活实验 |
实验开始前一天,将 Calu-6 细胞以每孔 1 × 10 4 细胞的密度接种在 384 孔板中,在补充有 10% 胎牛的 MEM 中生长血清和1%青霉素/链霉素。根据制造商的说明使用 Proteasome-Glo 测定试剂,通过跟踪胰凝乳蛋白酶样底物 Suc-LLVY-氨基荧光素在荧光素酶存在下的水解来测量蛋白酶体活性。一种称为 LEADseeker 的装置用于测量光度。
26S蛋白酶体活性抑制实验: 1. 蛋白制备:通过亲和层析纯化重组人26S蛋白酶体,重悬于实验缓冲液(25 mM Tris-HCl,pH7.5,5 mM MgCl₂,1 mM DTT)[1] 2. 反应体系:100 μL反应混合物含26S蛋白酶体(0.2 μg)、荧光底物(β5亚基:Suc-LLVY-AMC;β1亚基:Z-nLPnLD-AMC;β2亚基:Z-ARR-AMC)及IXAZOMIB (MLN2238)(0.1 nM–1000 nM,溶剂为对照)[1] 3. 孵育与检测:37℃孵育90分钟,测定荧光强度(激发光380 nm,发射光460 nm)。抑制率=(1–药物组荧光强度/对照组荧光强度)×100%[1] 4. 数据分析:使用GraphPad Prism将抑制率拟合至四参数逻辑斯蒂曲线,计算IC₅₀值[1] |
| 细胞实验 |
实验开始前一天,使用在补充有 10% FBS 的 MEM 中培养的 Calu-6 细胞,将 1 × 10 4 细胞接种到 384 孔板的每个孔中, 1%青霉素/链霉素。将细胞在 37°C 下用不同剂量的 MLN2238 或硼酸佐米(0.5% 最终 v/v DMSO)处理一小时,以进行 IC50 计算。为了进行可逆性测试,将细胞在 37°C 下暴露于 1 μM Bortezomib 或 MLN2238 中 30 分钟。处理后,细胞在培养基中洗涤三次以消除硼替佐米或 MLN2238。在 37°C 下再孵育 4 小时后,从细胞中除去培养基并更换为新培养基。
MTT抗增殖实验(文献[1]、[2]): 1. 细胞接种:MM/MCL细胞以5×10³个细胞/孔接种于96孔板,使用含10% FBS、1%青霉素-链霉素的RPMI 1640培养基[1][2] 2. 药物处理:加入IXAZOMIB (MLN2238)(0.1 nM–100 nM,每个浓度6个复孔),单独或与来那度胺(1 μM)联用,37℃、5% CO₂孵育72小时[1][2] 3. 活力检测:每孔加入20 μL MTT溶液(5 mg/mL PBS配制),孵育4小时。吸弃上清,加入150 μL DMSO溶解甲臜结晶,测定570 nm处吸光度,计算IC₅₀和联合指数(CI)[1][2] - 凋亡实验(Annexin V-FITC/PI法,文献[1]): 1. 细胞处理:RPMI 8226细胞(2×10⁵个细胞/孔,6孔板)用IXAZOMIB (MLN2238)(0 nM–20 nM)处理48小时[1] 2. 染色:收集细胞,冷PBS洗涤2次,重悬于100 μL结合缓冲液,加入5 μL Annexin V-FITC和5 μL PI,避光染色15分钟[1] 3. 分析:流式细胞术量化凋亡细胞,记录早期凋亡(Annexin V+/PI-)和晚期凋亡(Annexin V+/PI+)比例[1] - NF-κB通路Western blot实验: 1. 细胞处理:MM.1S细胞用0.5% FBS血清饥饿过夜,用IXAZOMIB (MLN2238)(0 nM–15 nM)处理24小时,再用TNF-α(10 ng/mL)刺激30分钟[1] 2. 裂解液制备:用含蛋白酶/磷酸酶抑制剂的RIPA缓冲液裂解细胞,BCA法测定蛋白浓度[1] 3. 免疫印迹:每泳道上样30 μg蛋白,经10% SDS-PAGE分离后转印至PVDF膜,用5%脱脂牛奶封闭1小时(室温),加入抗IκBα、抗p-p65(Ser536)及β-actin一抗(4℃过夜)。加入HRP偶联二抗(室温1小时),ECL化学发光检测信号[1] |
| 动物实验 |
小鼠:将新鲜解剖的CWR22肿瘤碎片(约20 mg)皮下注射到8至11周龄雄性CB17-SCID小鼠右侧背部。平均肿瘤体积(MTV)的计算公式为0.5×(长×宽²)。给药前,当MTV达到约150至200 mm³时,将动物随机分为治疗组(每组n=10)。通过计算研究结束时治疗组与对照组MTV的比值(T/C),确定抗肿瘤活性。
大鼠:向Sprague-Dawley大鼠静脉注射0.3或0.2 mg/kg的伊沙佐米(MLN2238)或0.2 mg/kg的硼替佐米,以确定这些药物在不同物种中的药代动力学特征。两种剂量的伊沙佐米(Ixazomib)的血浆暴露量(0.2 mg/kg 和 0.3 mg/kg 剂量的 AUC0-48h 分别为 704 和 1,070 h•ng/mL)均高于硼替佐米(Bortezomib)的 AUC0-48h(206 h•ng/mL),表明伊沙佐米(MLN2238)在啮齿动物体内的血浆暴露量也优于硼替佐米。 裸鼠 RPMI 8226 异种移植方案: 1. 动物饲养:雌性裸鼠(6-8 周龄,18-22 g)饲养于 SPF 级动物房(22-25°C,12 小时光照/黑暗循环),自由摄取食物和水[1] 2. 肿瘤植入:RPMI 8226 细胞(5×10⁶ 个细胞/只小鼠)将肿瘤细胞重悬于 100 μL PBS/Matrigel (1:1) 混合液中,皮下注射至小鼠右侧腹部 [1] 3. 分组和治疗:肿瘤体积达到约 100 mm³(第 0 天)时,随机分为 3 组。IXAZOMIB (MLN2238) 溶于 5% DMSO + 95% 生理盐水中,腹腔注射(10 μL/g 体重),剂量分别为 0.3 mg/kg 或 1 mg/kg,每日一次,连续 21 天。对照组仅注射溶剂 [1] 4. 监测和分析:每 3 天测量一次肿瘤体积(体积 = 长 × 宽² / 2);每周记录一次体重。小鼠采用 CO₂ 吸入法处死;切除肿瘤,称重,裂解后进行蛋白酶体活性测定(荧光底物法)[1] - 裸鼠 MM.1S 异种移植 + 来那度胺方案: 1. 肿瘤植入:将 MM.1S 细胞(2×10⁶ 个细胞/只小鼠)重悬于 100 μL PBS/Matrigel (1:1) 混合液中,皮下注射[2] 2. 治疗:伊沙唑米布 (MLN2238) 0.5 mg/kg(腹腔注射,每日一次)+ 来那度胺 25 mg/kg(灌胃,每日一次),持续 28 天(肿瘤体积约为 100 mm³ 时开始治疗)[2] 3. 分析:每 4 天测量一次肿瘤体积;通过 ELISA 法定量血清 M 蛋白;切除肿瘤进行 TUNEL 染色 [2] - 小鼠播散性多发性骨髓瘤模型: 1. 模型诱导:将 5TGM1 细胞(1×10⁶ 个细胞/只小鼠)静脉注射到 C57BL/KaLwRij 小鼠体内 [2] 2. 治疗:伊沙唑米布 (MLN2238) 0.75 mg/kg(腹腔注射,每日一次),持续 21 天(注射后 7 天开始)[2] 3. 分析:收集骨髓进行流式细胞术分析(CD138+ 细胞百分比);通过微型 CT 分析股骨的骨病变 [2] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
口服给药后,血浆药物浓度达峰时间为1小时。平均绝对口服生物利用度为58%。 尿液排泄率为62%,粪便排泄率为22%。 稳态分布容积为543升。 代谢/代谢物 预计伊沙佐米的代谢途径包括CYP和非CYP途径,且无明显的CYP同工酶贡献。在高于临床浓度时,伊沙佐米由多种 CYP 同工酶代谢,估计的相对贡献分别为 3A4 (42%)、1A2 (26%)、2B6 (16%)、2C8 (6%)、2D6 (5%)、2C19 (5%) 和 2C9 (<1%)。 生物半衰期 末端半衰期为 9.5 天。 小鼠腹腔注射药代动力学: 1. 药代动力学参数(0.5 mg/kg 腹腔注射剂量): - Cmax:~18 ng/mL (Tmax = 0.5 小时); - AUC₀-24h:~52 ng·h/mL; - 末端半衰期 (t₁/₂):~7.2 小时; - 清除率 (CL):~9.6 mL/min/kg [1] 2. 组织分布:腹腔注射 0.5 mg/kg 后 1 小时,RPMI 8226 肿瘤中 IXAZOMIB (MLN2238) 的浓度约为 42 ng/g,肿瘤/血浆浓度比约为 2.3 [1] |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
在伊沙佐米联合来那度胺和地塞米松的大型临床试验中,血清转氨酶水平升高较为常见,约10%的患者出现这种情况。然而,超过正常值上限5倍以上的数值较为罕见, 可能性评分:E(未经证实但怀疑是临床上明显的肝损伤的原因)。 妊娠和哺乳期影响 ◉ 哺乳期用药概述 目前尚无伊沙佐米在哺乳期临床应用的信息。由于其半衰期约为9.5天,因此可能在婴儿体内蓄积。此外,伊沙佐米还与来氟米特和地塞米松联合使用,这可能会增加婴儿的风险。制造商建议在接受伊沙佐米治疗期间以及末次给药后 90 天内停止母乳喂养。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对泌乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白质结合 99% 体外毒性: 1. 正常人骨髓基质细胞 (BMSC) 和外周血单核细胞 (PBMC):50 nM 伊沙佐米 (MLN2238)(72 小时处理)使细胞活力降低 <15%;未见明显细胞凋亡(Annexin V 染色)[2] - 体内毒性(文献[1],[2]): 1. 亚急性毒性(小鼠,1 mg/kg 腹腔注射,每日一次,持续 21 天): - 无明显体重减轻(<5% vs. 基线)或死亡; - 血清生化指标(ALT、AST、肌酐、BUN)均在正常范围内; - 肝脏、肾脏或心脏未见组织病理学病变[1] 2. 联合用药毒性(小鼠,IXAZOMIB (MLN2238) 0.5 mg/kg + 来那度胺 25 mg/kg,持续 28 天):与单药治疗相比,未见毒性增加;体重和器官功能未发生改变[2] - 血浆蛋白结合率:~99%(人血浆,37°C平衡透析)[1] |
| 参考文献 | |
| 其他信息 |
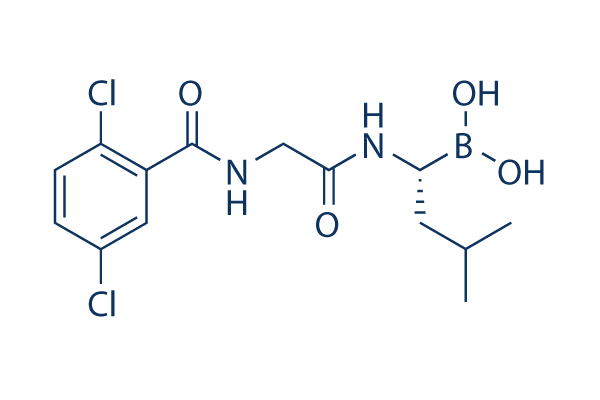
伊沙佐米是一种甘氨酸衍生物,它是N-(2,5-二氯苯甲酰基)甘氨酸的羧基与[(1R)-1-氨基-3-甲基丁基]硼酸的氨基缩合而成的酰胺。它是枸橼酸伊沙佐米的活性代谢物,用于多发性骨髓瘤的联合治疗。它具有多种功能,包括诱导细胞凋亡、作为孤儿药、蛋白酶体抑制剂、药物代谢物和抗肿瘤药物。它属于苯甲酰胺类、二氯苯类、甘氨酸衍生物和硼酸类化合物。
伊沙佐米是第二代蛋白酶体抑制剂 (PI),也是首个于2015年11月获得FDA批准的口服蛋白酶体抑制剂,用于治疗至少接受过一种既往治疗的多发性骨髓瘤患者,需与另外两种疗法(来那度胺和地塞米松)联合使用。研究发现,伊沙佐米在控制骨髓瘤生长和预防骨质流失方面与硼替佐米(首个获批用于多发性骨髓瘤治疗的蛋白酶体抑制剂)具有相似的疗效。伊沙佐米柠檬酸盐由武田制药以商品名Ninlaro上市销售,它是一种前体药物,给药后可迅速转化为其活性代谢物伊沙佐米。 伊沙佐米是一种蛋白酶体抑制剂。伊沙佐米的作用机制是作为蛋白酶体抑制剂。 伊沙佐米是一种小分子蛋白酶体抑制剂,与其他抗肿瘤药物联合用于治疗难治性多发性骨髓瘤。伊沙佐米治疗期间血清酶升高发生率较低,且临床上明显的急性肝损伤病例罕见。 伊沙佐米是MLN9708的活性代谢产物,MLN9708是一种第二代含硼肽类蛋白酶体抑制剂(PI),具有潜在的抗肿瘤活性。伊沙佐米与蛋白酶体的20S催化核心结合并抑制其活性,从而阻断蛋白酶体正常进行的靶向蛋白水解,导致不需要的或错误折叠的蛋白质积累;随后可能破坏多种细胞信号通路,最终诱导细胞凋亡。与第一代PI相比,第二代PI可能具有更优的药代动力学特性,效力更高,毒性更低。蛋白酶体是大型蛋白酶复合物,可降解不需要的或受损的泛素化蛋白质。 另见:枸橼酸伊沙佐米(其活性成分)。 药物适应症 伊沙佐米与来那度胺和地塞米松联合用于治疗至少接受过一种既往治疗的多发性骨髓瘤患者。 FDA标签 治疗淋巴系统恶性肿瘤(不包括多发性骨髓瘤),治疗多发性骨髓瘤 作用机制 伊沙佐米是一种N端加帽的二肽基亮氨酸硼酸,可逆性抑制20S蛋白酶体的CT-L蛋白水解(β5)位点。在高浓度下,伊沙佐米似乎还能抑制蛋白水解酶β1和β2亚基,并诱导泛素化蛋白的积累。 药效学 体外研究表明,伊沙佐米可诱导对其他常规疗法敏感或耐药的多发性骨髓瘤细胞凋亡。在小鼠异种移植模型中,伊沙佐米可抑制肿瘤生长。 作用机制:伊沙佐米(MLN2238)是口服前药枸橼酸伊沙佐米的活性形式。它选择性地与26S蛋白酶体的β5亚基结合,抑制胰凝乳蛋白酶样活性,阻断泛素化蛋白(例如IκBα、p53)的降解,并诱导癌细胞凋亡[1][2] - 临床意义:与口服前药不同,在临床前研究中,IXAZOMIB (MLN2238)是通过肠外途径(腹腔/静脉)给药的。它对硼替佐米耐药细胞的活性以及与来那度胺的协同作用支持其在复发/难治性多发性骨髓瘤中的应用[2] - 临床前研究重点:在皮下和播散性多发性骨髓瘤模型(模拟临床骨髓受累)中的疗效表明其具有治疗局部和全身性疾病的潜力[2] |
| 分子式 |
C14H19BCL2N2O4
|
|---|---|
| 分子量 |
361.03
|
| 精确质量 |
360.081
|
| 元素分析 |
C, 46.58; H, 5.30; B, 2.99; Cl, 19.64; N, 7.76; O, 17.73
|
| CAS号 |
1072833-77-2
|
| 相关CAS号 |
Ixazomib citrate;1239908-20-3
|
| PubChem CID |
25183872
|
| 外观&性状 |
Solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 折射率 |
1.546
|
| LogP |
2.82
|
| tPSA |
105.64
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
23
|
| 分子复杂度/Complexity |
412
|
| 定义原子立体中心数目 |
1
|
| SMILES |
ClC1C([H])=C([H])C(=C([H])C=1C(N([H])C([H])([H])C(N([H])[C@]([H])(B(O[H])O[H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H])=O)=O)Cl
|
| InChi Key |
MXAYKZJJDUDWDS-LBPRGKRZSA-N
|
| InChi Code |
InChI=1S/C14H19BCl2N2O4/c1-8(2)5-12(15(22)23)19-13(20)7-18-14(21)10-6-9(16)3-4-11(10)17/h3-4,6,8,12,22-23H,5,7H2,1-2H3,(H,18,21)(H,19,20)/t12-/m0/s1
|
| 化学名 |
[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]boronic acid
|
| 别名 |
MLN-2238; MLN2238; IXAZOMIB; MLN 2238; Trade name: Ninlaro
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.08 mg/mL (5.76 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 20.8 mg/mL澄清DMSO储备液加入400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.08 mg/mL (5.76 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 20.8 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.08 mg/mL (5.76 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 0.5% hydroxyethyl cellulose: 30 mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7699 mL | 13.8493 mL | 27.6985 mL | |
| 5 mM | 0.5540 mL | 2.7699 mL | 5.5397 mL | |
| 10 mM | 0.2770 mL | 1.3849 mL | 2.7699 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Daratumumab, Ixazomib, and Dexamethasone in AL Amyloidosis
CTID: NCT03283917
Phase: Phase 1 Status: Active, not recruiting
Date: 2024-08-15
|
|---|
|
|
|
|---|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
")
")
")
")
")
")
 463611831
463611831
