| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
T-type calcium channels (Caₐ₃.2): recombinant human Caₐ₃.2 (IC₅₀ = 2 μM, voltage-dependent block), rat dorsal root ganglion (DRG) neuron low voltage-activated (LVA) currents (IC₅₀ = 8 μM) [1]
Other calcium channels (Caₐ₁.2, Caₐ₂.2): IC₅₀ > 30 μM (significantly less active) [1] |
|---|---|
| 体外研究 (In Vitro) |
体外活性:ABT-639 是一种新型强效、外周作用、选择性 T 型 Ca2+ 通道阻滞剂,以电压依赖性方式阻断重组人 T 型 (Cav3.2) Ca2+ 通道 (IC50=2 μM) 并减弱大鼠 DRG 神经元中的低电压激活 (LVA) 电流 (IC50=8 μM)。 ABT-639 在其他 Ca2+ 通道(例如 Cav1.2 和 Cav2.2)上的活性明显较低(IC50>30 mM)。口服给药后,ABT-639 在大鼠膝关节疼痛模型中产生剂量依赖性镇痛作用(ED₅₀ = 2 mg/kg,口服)。 ABT-639(10-100 mg/kg,口服)还增加了多种神经性疼痛模型(例如脊神经结扎、CCI 和长春新碱诱导)的触觉异常性疼痛阈值。 [更正]。 ABT-639 不会减弱弗氏完全佐剂或角叉菜胶诱导的炎性疼痛模型中的痛觉过敏。在较高剂量(例如100-300 mg/kg)下,ABT-639 并未显着改变血流动力学或精神运动功能。 ABT-639 的镇痛作用为外周 T 型 (Ca(v)3.2) 通道在慢性疼痛状态中的作用提供了新的见解。激酶测定:细胞测定:ABT-639 以电压依赖性方式阻断重组人 T 型 (Cav3.2) Ca2+ 通道 (IC50=2 μM) 并减弱大鼠 DRG 神经元中的低电压激活 (LVA) 电流 (IC50) =8μM)。 ABT-639 在其他 Ca2+ 通道(例如 Cav1.2 和 Cav2.2)上的活性明显较低(IC50>30 mM)。 ABT-639 在啮齿类动物中具有高口服生物利用度 (%F=73)、低蛋白结合 (88.9%) 和低脑:血浆比 (0.05:1)。口服给药后
1. 通道阻断活性:ABT-639 以电压依赖性方式阻断重组人源T型(Caₐ₃.2)钙通道,IC₅₀值为2 μM;可抑制大鼠背根神经节(DRG)神经元的低电压激活(LVA)钙电流,IC₅₀值为8 μM;该化合物对其他钙通道亚型(Caₐ₁.2、Caₐ₂.2)的活性显著降低,IC₅₀值均大于30 μM,体现出对T型(Caₐ₃.2)通道的高选择性[1] |
| 体内研究 (In Vivo) |
ABT-639 减弱大鼠 DRG 神经元中的低电压激活 (LVA) 电流 (IC50=8 μM),并以电压依赖性方式阻断重组人 T 型 (Cav3.2) Ca2+ 通道 (IC50=2 μM)。 ABT -639 在其他 Ca2+ 通道 (IC50>30 mM)(例如 Cav1.2 和 Cav2.2)上表现出显着较低的活性。在啮齿动物中,ABT-639 表现出低蛋白结合率 (88.9%)、低脑:血浆比率 (0.05:1) 和高口服生物利用度 (%F=73)。在膝关节疼痛大鼠模型中,口服 ABT-639 会产生剂量依赖性镇痛作用(ED50=2 mg/kg,口服)。在各种神经性疼痛模型中,例如脊神经结扎、CCI、长春新碱诱导的和辣椒素继发性超敏反应,ABT-639(10-100 mg/kg,口服)也会提高触觉异常性疼痛阈值。在角叉菜胶或弗氏完全佐剂诱导的炎性疼痛模型中,ABT-639 不会减轻痛觉过敏。较高剂量的 ABT-639(例如 100-300 mg/kg)对血流动力学或精神运动功能没有明显影响。 ABT-639 的镇痛特性为慢性疼痛状态下外周 T 型 (Cav3.2) 通道的功能提供了新的见解[1]。
1. 伤害性疼痛模型中的镇痛作用:大鼠膝关节疼痛模型中,口服给予ABT-639 可产生剂量依赖性镇痛效果,ED₅₀值为2 mg/kg(口服)[1] 2. 神经性疼痛模型中的作用:ABT-639(10-100 mg/kg,口服)可提高多种大鼠神经性疼痛模型(脊神经结扎、慢性压迫性损伤(CCI)、长春新碱诱导的神经性疼痛)的触觉异常性疼痛阈值[1] 3. 炎症性疼痛模型中的作用:ABT-639 无法缓解完全弗氏佐剂或角叉菜胶诱导的大鼠炎症性疼痛模型中的痛觉过敏[1] 4. 血流动力学和精神运动影响:高剂量(100-300 mg/kg,口服)ABT-639 未显著改变大鼠的血流动力学(如血压、心率)或精神运动功能[1] |
| 酶活实验 |
1. 重组人源Caₐ₃.2通道活性实验:将重组人源Caₐ₃.2 T型钙通道制备物暴露于不同浓度的ABT-639,并设置不同电压条件;检测通道阻断活性,确定IC₅₀值(2 μM),验证ABT-639 的电压依赖性阻断特征[1]
2. 大鼠DRG神经元LVA电流实验:分离并培养原代大鼠DRG神经元,用不同浓度的ABT-639 处理;采用膜片钳技术记录LVA钙电流,定量ABT-639 对该电流的抑制作用,计算IC₅₀值(8 μM)[1] 3. 其他钙通道活性实验:将Caₐ₁.2、Caₐ₂.2钙通道制备物用ABT-639(浓度高于30 μM)处理;检测通道活性,确定IC₅₀值(>30 μM),证实ABT-639 对这些亚型的低活性[1] |
| 细胞实验 |
1. 大鼠DRG神经元培养及LVA电流检测:分离、纯化大鼠DRG神经元并在适宜培养基中培养;待神经元达到所需状态后,与不同浓度的ABT-639 共孵育;采用膜片钳电生理技术检测低电压激活钙电流,评估ABT-639 对该电流的抑制作用,确定其对大鼠DRG神经元LVA电流的IC₅₀值[1]
|
| 动物实验 |
10-100 mg/kg,口服
大鼠膝关节疼痛模型 1. 大鼠膝关节疼痛模型(伤害性疼痛):采用雄性Sprague-Dawley大鼠建立膝关节疼痛模型;口服不同剂量的ABT-639,并评估其镇痛效果以确定ED₅₀值(2 mg/kg,口服)[1] 2. 神经病理性疼痛模型(脊神经结扎、CCI、长春新碱诱导):采用雄性Sprague-Dawley大鼠建立相应的神经病理性疼痛模型; ABT-639以10-100 mg/kg的剂量口服给药,并在特定时间点测量触觉异常性疼痛阈值以评估治疗效果[1] 3.炎症性疼痛模型(弗氏完全佐剂/角叉菜胶诱导):雄性Sprague-Dawley大鼠注射弗氏完全佐剂或角叉菜胶以诱导炎症性疼痛;ABT-639口服给药,并评估痛觉过敏以确认ABT-639对炎症性疼痛无影响[1] 4.血流动力学/精神运动功能评估:雄性Sprague-Dawley大鼠口服高剂量ABT-639(100-300 mg/kg);在特定时间段内监测血流动力学参数(例如血压、心率)和精神运动功能,以评估潜在的不良反应[1] 5. 啮齿动物药代动力学评估:对啮齿动物(未指定具体品系)口服ABT-639;在不同时间点采集血浆和脑组织样本,并测量药物浓度以确定口服生物利用度、血浆蛋白结合率和脑/血浆比值[1] |
| 药代性质 (ADME/PK) |
1. 口服生物利用度:ABT-639在啮齿动物中具有较高的口服生物利用度,%F值为73%[1]
2. 血浆蛋白结合率:ABT-639在啮齿动物中的血浆蛋白结合率为88.9%(蛋白结合率低)[1] 3. 组织分布:ABT-639在啮齿动物中显示出较低的脑/血浆比值(0.05:1),表明其穿过血脑屏障的能力有限,且具有外周选择性[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
1. 血流动力学/精神运动毒性:在剂量高达 300 mg/kg(口服)的情况下,ABT-639 未引起大鼠血流动力学参数(例如血压、心率)或精神运动功能的显著改变,表明其对这些终点指标无急性毒性[1]
2. 血浆蛋白结合率:ABT-639 的血浆蛋白结合率为 88.9%(啮齿动物),未见报道与蛋白结合置换相关的药物相互作用[1] |
| 参考文献 | |
| 其他信息 |
ABT-639 正在进行临床试验 NCT01345045(一项多中心研究,比较 ABT-639 与安慰剂在糖尿病神经性疼痛患者中的镇痛效果和安全性)。
T 型钙通道阻滞剂 ABT-639 是一种口服生物利用度高的 CaV3.2 T 型钙通道阻滞剂,具有潜在的抗痛觉过敏活性。口服后,ABT-639 可选择性地结合并阻断位于外周感觉神经元中的低电压门控 T 型钙通道的 CaV3.2 亚型。这可阻止膜去极化时钙离子流入细胞。抑制神经元过度兴奋和伤害性外周感觉神经元的放电,从而产生镇痛作用。 CaV3.2 T型通道的表达在伤害性疼痛和神经性疼痛中起着关键作用。 1. 作用机制:ABT-639是一种外周作用的选择性T型(Caₐ₃.2)钙通道阻滞剂;T型Ca²⁺通道的激活通过促进神经元过度兴奋期间的动作电位爆发和膜电位调节来促进伤害性信号传导,而ABT-639通过阻断Caₐ₃.2通道来减弱这种信号传导[1] 2. 治疗特性:ABT-639能有效减轻大鼠的伤害性疼痛(膝关节疼痛)和神经性疼痛,但对炎症性疼痛无效,这突显了其对特定疼痛状态的选择性疗效;其外周选择性(脑渗透性低)以及高剂量下无血流动力学/精神运动毒性,支持其作为慢性疼痛安全治疗药物的潜力[1] 3. 背景:基因敲低研究支持T型Ca²⁺通道(Caₐ₃.2)在慢性疼痛中的作用,这些研究表明DRG神经元中的LVA电流降低,CCI模型中的神经性疼痛减轻;ABT-639是一种新型的靶向该通道亚型的外周作用阻滞剂[1] |
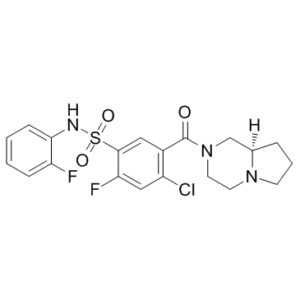
| 分子式 |
C20H20CLF2N3O3S
|
|
|---|---|---|
| 分子量 |
455.91
|
|
| 精确质量 |
455.088
|
|
| CAS号 |
1235560-28-7
|
|
| 相关CAS号 |
ABT-639 hydrochloride;1235560-31-2
|
|
| PubChem CID |
46851313
|
|
| 外观&性状 |
White to off-white solid powder
|
|
| 密度 |
1.5±0.1 g/cm3
|
|
| 沸点 |
612.2±65.0 °C at 760 mmHg
|
|
| 闪点 |
324.0±34.3 °C
|
|
| 蒸汽压 |
0.0±1.8 mmHg at 25°C
|
|
| 折射率 |
1.653
|
|
| LogP |
2.6
|
|
| tPSA |
78.1
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
7
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
30
|
|
| 分子复杂度/Complexity |
737
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
C1C[C@@H]2CN(CCN2C1)C(=O)C3=CC(=C(C=C3Cl)F)S(=O)(=O)NC4=CC=CC=C4F
|
|
| InChi Key |
AGPIHNZOZNKRGT-CYBMUJFWSA-N
|
|
| InChi Code |
InChI=1S/C20H20ClF2N3O3S/c21-15-11-17(23)19(30(28,29)24-18-6-2-1-5-16(18)22)10-14(15)20(27)26-9-8-25-7-3-4-13(25)12-26/h1-2,5-6,10-11,13,24H,3-4,7-9,12H2/t13-/m1/s1
|
|
| 化学名 |
5-[(8aR)-3,4,6,7,8,8a-hexahydro-1H-pyrrolo[1,2-a]pyrazine-2-carbonyl]-4-chloro-2-fluoro-N-(2-fluorophenyl)benzenesulfonamide
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1934 mL | 10.9671 mL | 21.9342 mL | |
| 5 mM | 0.4387 mL | 2.1934 mL | 4.3868 mL | |
| 10 mM | 0.2193 mL | 1.0967 mL | 2.1934 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 GV-58
GV-58
 丁酸氯维地平
丁酸氯维地平
 NNC 55-0396
NNC 55-0396
 依碳氯替泼诺
依碳氯替泼诺
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
