| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
GABAA Receptor (α₁β₂γ₂s subtype): Imperatorin potentiates GABA-induced chloride ion current through this receptor. (At 100 μM, potentiation = 50.5 ± 16.3%; at 300 μM, potentiation = 109.8 ± 37.7%) [1]
- Benzodiazepine Site of GABAA Receptor: Imperatorin inhibits [³H]diazepam binding. (IC50 = 12.3 μM) [1] - GABA Transaminase (GABA-T): Imperatorin irreversibly inhibits GABA-T in a time- and concentration-dependent manner. (No IC50 reported) [1] - Acetylcholinesterase (AChE): Imperatorin inhibits AChE. (Minimum inhibitory quantity on TLC: 0.1 μg; at 100 μg/mL, inhibition = 13.75-46.11%) [1] - Butyrylcholinesterase (BChE): Imperatorin selectively inhibits BChE. (IC50 = 31.4 μM; at 100 μg/mL, inhibition = 37.46-83.98%) [1] - β-Secretase (BACE-1): Imperatorin inhibits BACE-1. (IC50 = 91.8 μM) [1] - Cyclooxygenase-1 (COX-1) and Cyclooxygenase-2 (COX-2): Imperatorin inhibits both isoforms. (COX-1/COX-2 selectivity ratio = 0.90 at 100 μM) [1] - 5-Lipoxygenase (5-LOX): Imperatorin inhibits 5-LOX. (IC50 < 15 μM) [1] - Nitric Oxide Synthase (NOS): Imperatorin inhibits NO synthesis. (IC50 = 9.2 μM) [1] - Cytochrome P450 1A1 (CYP1A1): Imperatorin inhibits ethoxyresorufin O-dealkylase (EROD) activity mediated by CYP1A1 in a competitive manner. [1] |
|---|---|
| 体外研究 (In Vitro) |
异黄酮是一种植物次生代谢产物,属于香豆素家族,具体来说是呋喃香豆素类。异黄酮通过α1β2η2S受体发挥作用,增加GABA诱导的氯离子电流(IGABA)。浓度分别为100 μmol和300 μmol时,异黄酮可使IGABA增加50.5±16.3%和109.8±37.7%。体外实验表明,[3H]地西泮与大鼠脑内GABAA受体苯二氮卓类结合位点的结合可被白藜芦醇根中存在的异黄酮和异黄酮抑制。异黄酮的IC50为12.3 μmol,而异黄酮的IC50为400 nmol。欧前胡素通过与GABA-T活性位点永久结合,在3.5至14 mmol的浓度范围内,以浓度依赖性和时间依赖性的方式,强效且不可逆地抑制GABA-T。欧前胡素是一种剂量依赖性的可逆性乙酰胆碱酯酶(AChE)抑制剂。本研究采用分光光度法,检测了浓度分别为12.5、25、50和100 μg/mL的欧前胡素和当归果实粗提物对AChE和丁酰胆碱酯酶(BChE)的抑制活性。结果表明,欧前胡素对AChE的抑制率较低(13.75-46.11%),而对BChE的抑制率较高(37.46-83.98%)。BChE的IC50值为31.4 μmol,表明欧前胡素对BChE具有选择性,而对AChE无选择性。研究发现,最有效的BACE-1抑制剂是异黄酮和(+)-白芷酚,其IC50值分别为91.8和104.9 μmol。异黄酮还表现出强烈的NO合成抑制作用(IC50=9.2 μmol)[1]。异黄酮的EC50为12.6±3.2 μM,是TRPV1的弱激动剂,TRPV1是一种与感知一系列不愉快刺激相关的通道[2]。
GABAA受体调节:在表达α₁β₂γ₂s GABAA受体的非洲爪蟾卵母细胞中,异黄酮(100-300 μM)呈剂量依赖性地增强GABA诱导的氯离子电流。 [1] - 苯二氮卓受体结合:异戊二烯酮抑制[³H]地西泮与大鼠脑GABAA受体苯二氮卓结合位点的结合,IC50值为12.3 μM。[1] - GABA-T抑制:异戊二烯酮(3.5-14 mM)通过与酶的活性位点结合,以时间和浓度依赖的方式显著且不可逆地抑制GABA-T。[1] - 乙酰胆碱酯酶(AChE)和丁酰胆碱酯酶(BChE)抑制:测试了异戊二烯酮对乙酰胆碱酯酶和丁酰胆碱酯酶的抑制作用。结果表明,异戊二烯酮对BChE的选择性高于AChE(IC50 = 31.4 μM)。在薄层色谱生物自显影法中,AChE的最低抑制量为0.1 μg。在浓度为 100 μg/mL 的分光光度法测定中,异戊二烯酮对乙酰胆碱酯酶 (AChE) 的抑制率为 13.75-46.11%,对丁酰胆碱酯酶 (BChE) 的抑制率为 37.46-83.98%。分子对接研究证实了异戊二烯酮与 BChE 活性谷的相互作用。[1] - BACE-1 抑制:异戊二烯酮对 β-分泌酶 (BACE-1) 的抑制 IC50 为 91.8 μM。其代谢产物黄毒醇的 IC50 为 185.6 ± 6.8 μM。[1] - 血管舒张:异戊二烯酮可诱导用氯化钾或内皮素-1 预收缩的离体大鼠肠系膜动脉以及用去甲肾上腺素预收缩的人大网膜动脉的血管舒张。该作用与内皮细胞无关。在小鼠胸主动脉中,血管舒张的IC50值为12.2 μmol/L(异异戊二烯酮为47.6 μmol/L),在兔阴茎海绵体中为0.85 mmol/L。[1] - 抗菌活性:异戊二烯酮对多种细菌具有抗菌活性。MIC值:蜡样芽孢杆菌(5 × 10⁻⁵ μg/mL)、霍乱弧菌(8 μg/mL)、金黄色葡萄球菌(32 μg/mL)、枯草芽孢杆菌(0.125-500 μg/mL,取决于研究)、阴沟肠杆菌(0.028 mg/mL)、肺炎克雷伯菌(0.030 mg/mL)、变形链球菌(0.018 mg/mL)、草绿色链球菌(0.015 mg/mL)。它对白色念珠菌的活性较弱(MIC = 0.25 mg/mL)。[1] - 抗炎活性:异戊二烯酮抑制离子载体刺激的小鼠腹腔巨噬细胞中 PGE₂ 和 LTC₄ 的释放。它还能抑制 LPS 诱导的 PGE₂ 释放(浓度为 25-100 μM 时,PGE₂ 浓度从 1.13 ng/mL 降至 0.13 ng/mL)和 NO 合成(IC50 = 9.2 μM)。[1] - 抗癌活性(诱导细胞凋亡):在 HeLa 细胞中,异戊二烯酮(50-200 μM)可诱导细胞凋亡,并增加 caspase-3 和 caspase-8 的活性。在HL-60细胞中,异戊二烯酮(0.1-50 μM)以剂量依赖的方式降低细胞活力(50 μM时从100%降至28%),诱导caspase-3和caspase-9活化(但不诱导caspase-8活化),并导致细胞核碎裂,表明其作用途径涉及线粒体。在HepG2细胞中,异戊二烯酮(30-90 μM)降低Bcl-2表达,增加Bax、Bad、tBid和p53表达,同时降低pro-caspase-3、-8和-9水平。[1] - 抗癌活性(自噬诱导):在HeLa细胞中,异戊二烯酮(50-200 μM)可诱导自噬,表现为LC3裂解和热休克蛋白(尤其是Hsp27)表达抑制。仅在极高浓度(200 μM)下观察到细胞坏死。 [1] - 抗癌活性(DNA加合物抑制):在MCF-7细胞中,异戊二烯酮(10 μM)分别抑制了DMBA和B[a]P DNA加合物的形成58%和63%。[1] - 抗病毒活性(HIV-1复制):在HeLa、MT-2和Jurkat T细胞中,异戊二烯酮(10-100 μM,24-72 h)通过阻断Sp1依赖性转录和抑制细胞周期蛋白D1的表达来抑制HIV-1复制,导致G1期细胞周期阻滞。[1] - 抗骨质疏松活性:在成骨细胞培养物中,与对照组相比,异戊二烯酮(10 μM)使BMP-2基因表达增加3倍以上,胶原蛋白含量增加2倍以上,矿化结节形成增加约225%。 [1] - 肌肉松弛活性:在离体大鼠空肠条中,异戊二烯酮(0.001-100 μM)可引起肌肉逐渐松弛,其在 25-50 μM 时的效果与异丙肾上腺素(0.1 μM)相当。[1] - 酶诱导(I 期和 II 期):异戊二烯酮可诱导 I 期(细胞色素 P450)和 II 期(谷胱甘肽 S-转移酶)酶的表达。使用 CDNB、DCNB 和 EA 底物时,GST 的最大诱导率分别为对照组的 260%、300% 和 170%。 [1] - 致癌物解毒酶诱导:异戊烯基腺嘌呤(6.25-200 μM,24 小时)可诱导 NQO1 活性从 9.5 nmol/min/mg 蛋白增加至 13 nmol/min/mg 蛋白,GST 活性从 10.9 nmol/min/mg 蛋白增加至 34.54 nmol/min/mg 蛋白。[1] |
| 体内研究 (In Vivo) |
异戊烯基腺苷在注射后30分钟,剂量为10和20 mg/kg时,具有抗焦虑作用并能增强记忆力。它还能促进学习的两个不同阶段:习得和巩固。研究还表明,急性给予异戊烯基腺苷(剂量为10和20 mg/kg)可减轻尼古丁(皮下注射,0.1 mg/kg)的致焦虑作用。腹腔注射异戊烯基腺苷(剂量为30和40 mg/kg)可显著增强卡马西平对最大电击诱发癫痫发作的抗惊厥作用。这使得卡马西平的ED50分别从10.8 mg/kg降低至6.8 mg/kg(降低34%)和6 mg/kg(降低42%)。此外,当给予30 mg/kg的异戊二烯酮和6.8 mg/kg的卡马西平时,脑内卡马西平的总浓度增加了85%(从1.260 μg/mL增至2.328 μg/mL)。这种增加可能是由于血脑屏障通透性改变或类似于多药耐药蛋白抑制剂的作用所致[1]。异戊二烯酮是一种天然存在的呋喃香豆素,可抑制乙酰胆碱酯酶的活性并使γ-氨基丁酸转氨酶失活。在注射东莨菪碱(1 mg/kg)前急性给予5 mg/kg和10 mg/kg的异戊二烯酮,可改善东莨菪碱引起的记忆巩固和习得障碍。此外,连续7天每日两次给予最大剂量(10 mg/kg)的异戊二烯酮,可显著降低东莨菪碱对记忆习得的影响。相比之下,呋喃香豆素的剂量为 5 和 10 mg/kg 时具有相同的效果。当记忆力改善时,效果显著。测量指标为记忆巩固 [3]。
记忆增强:在 10 月龄小鼠中,口服异戊二烯酮(0.79 mg/kg,持续 14 天或更长时间)显著延长了被动回避测试中的下台阶潜伏期。[1] - 抗焦虑和认知效应:在瑞士雄性小鼠中,腹腔注射异戊二烯酮(10 和 20 mg/kg,注射后 30 分钟)显示出抗焦虑作用,并改善了高架十字迷宫和被动回避测试中的记忆获得和巩固。[1] - 逆转尼古丁诱导的变化:异戊二烯酮(1 mg/kg,腹腔注射,每日两次,持续 6 天)逆转了小鼠尼古丁诱导的焦虑、认知障碍和氧化应激,降低了 MDA 水平,并提高了 GPx、SOD 和 GR 的活性。 [1] - 东莨菪碱诱导的记忆障碍逆转:异戊二烯酮(5 和 10 mg/kg,腹腔注射)可改善东莨菪碱(1 mg/kg)引起的记忆获取和巩固障碍。重复给药(10 mg/kg,每日两次,持续 7 天)也可减轻东莨菪碱诱导的记忆缺陷和氧化应激。[1] - 抗惊厥活性:在小鼠最大电休克癫痫(MES)模型中,异戊二烯酮(10-100 mg/kg,腹腔注射)可提高癫痫阈值。最佳效果出现在注射后 30 分钟,100 mg/kg 可使阈值提高 68%。ED50 值范围为 167-290 mg/kg。神经毒性 TD50 值范围为 329-443 mg/kg。 [1] - 协同抗惊厥作用:异戊二烯酮(30-40 mg/kg,腹腔注射)可增强卡马西平(ED50 降低 34-42%)、苯妥英钠(ED50 降低 34%)和苯巴比妥(ED50 降低 38%)的抗惊厥活性。该组合不损害长期记忆、运动协调性或握力。[1] - 抗高血压作用:在肾性高血压大鼠中,异戊二烯酮(6.25、12.5 和 25 mg/kg/天,灌胃)可降低血压,减少脂质过氧化物水平,并通过下调 NADPH 氧化酶的表达来提高抗氧化酶活性。 [1] - 抑制心脏肥大:在高血压大鼠中,欧前胡素(25 mg/kg/天,持续4周)可降低心肌细胞直径(低剂量组从51 μm降至45 μm,高剂量组降至31 μm),减少心肌纤维化4-12%,并改善血流动力学参数。[1] - 抗肿瘤活性:在荷HepG2裸鼠中,口服欧前胡素(50和100 mg/kg/天,持续14天)可显著抑制肿瘤生长,抑制率分别为31.93%和63.18%,且无明显副作用(体重减轻、心脏毒性、肝毒性)。 [1] - 保肝活性:在刀豆蛋白A诱导或抗Fas抗体诱导的小鼠肝炎模型中,异戊二烯酮(100 mg/kg)可抑制83%的血浆丙氨酸氨基转移酶(ALT)活性升高。[1] |
| 酶活实验 |
GABA-T抑制试验:体外评价了异戊二烯对GABA转氨酶(GABA-T)的抑制作用。试验结果表明,异戊二烯(3.5-14 mM)通过与活性位点不可逆结合,以时间和浓度依赖的方式抑制该酶的活性。[1]
- AChE和BChE抑制试验:采用改良的Ellman法评价了异戊二烯对乙酰胆碱酯酶和丁酰胆碱酯酶的抑制活性。对于薄层色谱生物自显影法,将异戊二烯溶液点样于薄层色谱板上,显色后,喷洒酶和底物溶液以检测白色抑制斑点。对于分光光度法,将不同浓度的异戊二烯(12.5、25、50和100 μg/mL)与酶和底物混合孵育,测定吸光度以计算抑制率。 [1] - BACE-1抑制试验:在异戊二烯存在下,通过测量酶对特定荧光底物的裂解来评估β-分泌酶的抑制活性。IC50值由浓度-反应曲线计算得出。[1] - 血管舒张试验:将离体动脉置于组织浴槽中,并用血管收缩剂(苯肾上腺素、氯化钾、内皮素-1、去甲肾上腺素)预收缩。加入累积浓度的异戊二烯,并测量等长张力以评估舒张作用。通过比较完整和去除内皮的内皮制备物来评估内皮依赖性。[1] - 抗菌试验:使用刃天青或其他细菌生长指示剂的微量稀释法或肉汤稀释法测定MIC值。 [1] - 抗炎活性检测:用离子载体或LPS刺激小鼠腹腔巨噬细胞。通过免疫测定法检测PGE₂和LTC₄的释放。通过Griess反应检测NO的产生。[1] - 细胞凋亡检测:通过MTT法评估细胞活力。通过DNA片段化分析(电泳)、caspase活性测定(使用荧光底物)和凋亡相关蛋白(Bcl-2、Bax、Bad、tBid、p53、caspase)的Western blot分析检测细胞凋亡。[1] - 自噬活性检测:通过LC3裂解(Western blot)和热休克蛋白表达抑制来评估自噬。[1] - I期和II期酶诱导活性检测:制备用异戊二烯酮处理的小鼠的肝微粒体。 CYP1A1活性采用乙氧基试卤灵O-脱烷基酶(EROD)法测定。GST活性采用CDNB、DCNB和EA作为底物测定。[1] |
| 细胞实验 |
非洲爪蟾卵母细胞:将编码 α₁β₂γ₂s GABAA 受体亚基的 cRNA 注射到非洲爪蟾卵母细胞中。1-3 天后,进行双电极电压钳记录,以测量 GABA 诱导的电流和异戊二烯酮的作用。[1] - HeLa 细胞:培养人宫颈癌细胞,并用异戊二烯酮(50-200 μM)处理,同时或不添加顺铂。评估细胞活力、凋亡、caspase 活性和自噬标志物。[1] - HL-60 细胞:用异戊二烯酮(0.1-50 μM)处理人早幼粒白血病细胞。检测细胞活力、caspase-3、-8 和 -9 活性以及核形态。 [1] - HepG2 细胞:用异戊二烯酮(30-90 μM)处理人肝癌细胞。进行蛋白质印迹分析,检测 Bcl-2、Bax、Bad、tBid、p53 和 caspase 的表达。[1] - MCF-7 细胞:用异戊二烯酮(10 μM)处理人乳腺癌细胞,以评估其对 DMBA 和 B[a]P DNA 加合物形成的抑制作用。[1] - HIV-1 复制细胞系:用异戊二烯酮(10-100 μM)处理 HeLa、MT-2(人 T 细胞白血病)和 Jurkat T 细胞,并感染 HIV-1 假型病毒。分析病毒复制和细胞周期进程。[1] - 成骨细胞培养:用异戊二烯酮(10 μM)处理细胞,并检测 BMP-2 基因表达、胶原含量和矿化结节形成。 [1]
|
| 动物实验 |
小鼠记忆研究:** 10月龄小鼠口服给予欧前胡素(0.79 mg/kg)或欧当归提取物,持续14天或更长时间。采用下台阶被动回避测试评估记忆力。[1]
- **小鼠抗焦虑/认知研究:** 瑞士雄性小鼠在进行高架十字迷宫、改良高架十字迷宫和被动回避测试前30分钟腹腔注射欧前胡素(1、5、10、20 mg/kg)。[1] - **小鼠氧化应激研究:** 小鼠腹腔注射欧前胡素(1 mg/kg,每日两次,持续6天)和尼古丁(0.1 mg/kg,皮下注射)。分析脑组织中丙二醛(MDA)、谷胱甘肽过氧化物酶(GPx)、超氧化物歧化酶(SOD)和谷胱甘肽还原酶(GR)的水平。 [1] - **小鼠东莨菪碱研究:**小鼠在接受东莨菪碱(1 mg/kg)前,腹腔注射异戊二烯酮(5 或 10 mg/kg)。通过被动回避实验评估记忆力,并测量氧化应激标志物。[1] - **小鼠抗惊厥研究:**小鼠在最大电休克惊厥(MES)试验前不同时间点腹腔注射异戊二烯酮(10-100 mg/kg)。测定惊厥阈值,并计算 ED50 和 TD50 值。[1] - **大鼠抗高血压研究:**肾性高血压大鼠接受胃内灌注异戊二烯酮(6.25、12.5 或 25 mg/kg/天)。测量血压,并分析肾组织中 NADPH 氧化酶的表达和抗氧化标志物。 [1] - **大鼠心脏肥大研究:**高血压大鼠接受异戊二烯酮(25 mg/kg/天,灌胃)。检测心肌组织,评估心肌细胞直径、纤维化程度和血流动力学参数。[1] - **小鼠异种移植研究:**荷HepG2肿瘤的裸鼠接受异戊二烯酮(50或100 mg/kg/天,口服)治疗14天。监测肿瘤体积、体重和毒性反应。[1] - **小鼠肝炎研究:**用刀豆蛋白A或抗Fas抗体诱导肝炎的小鼠接受异戊二烯酮(100 mg/kg)治疗。测定血浆ALT活性。[1] 小鼠记忆研究:10月龄小鼠口服异戊二烯酮(0.79 mg/kg)或欧当归提取物,持续14天或更长时间。采用下台阶被动回避测试评估记忆力。[1] - 小鼠抗焦虑/认知研究:瑞士雄性小鼠在进行高架十字迷宫、改良高架十字迷宫和被动回避测试前30分钟,腹腔注射异戊二烯酮(1、5、10、20 mg/kg)。[1] - 小鼠氧化应激研究:小鼠腹腔注射异戊二烯酮(1 mg/kg,每日两次,连续6天)和尼古丁(0.1 mg/kg,皮下注射)。分析脑组织中MDA、GPx、SOD和GR的水平。[1] - 小鼠东莨菪碱研究:小鼠在注射东莨菪碱(1 mg/kg)前,腹腔注射异戊二烯酮(5或10 mg/kg)。采用被动回避测试评估记忆力,并测量氧化应激标志物。 [1] - 小鼠抗惊厥研究:小鼠在最大电休克惊厥(MES)试验前不同时间点接受异戊二烯酮(10-100 mg/kg,腹腔注射)。测定惊厥阈值,并计算ED50和TD50值。[1] - 大鼠抗高血压研究:肾性高血压大鼠接受异戊二烯酮(6.25、12.5或25 mg/kg/天,灌胃给药)。测量血压,并分析肾组织中NADPH氧化酶的表达和抗氧化标志物。[1] - 大鼠心脏肥大研究:高血压大鼠接受异戊二烯酮(25 mg/kg/天,灌胃给药)。检测心脏组织中心肌细胞直径、纤维化程度和血流动力学参数。 [1] - 小鼠异种移植研究:荷瘤裸鼠(HepG2肿瘤)接受异戊烯基腺苷(50或100 mg/kg/天,口服)治疗14天。监测肿瘤体积、体重和毒性反应。[1] - 小鼠肝炎研究:用刀豆蛋白A或抗Fas抗体诱导肝炎的小鼠接受异戊烯基腺苷(100 mg/kg)治疗。检测血浆ALT活性。[1] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
我们开发了一种快速灵敏的检测方法,用于定量测定血浆和组织中的异戊二烯酮。采用气相色谱/质谱联用(GC/MS)技术,在选择离子监测模式下进行分析。获得的主要药代动力学参数为:Tmax = 1.23 ± 0.26 h,Cmax = 0.95 ± 0.38 μg/mL,AUC = 3.42 ± 0.52 h·μg/mL,Ka = 1.34 ± 0.18 h。结果表明,异戊二烯酮易于吸收,但口服给药后3至12小时内消除缓慢。大鼠肝脏、肾脏、肺和心脏中的异戊二烯酮浓度高于其他器官。为了测定血清中的游离异戊二烯酮,使用截留分子量为10 kDa的超滤膜过滤样品,并采用液液萃取法进行提取。大鼠血浆、自发性高血压大鼠血浆、人血浆和人血清白蛋白中的蛋白结合率分别为84±3%、69±7%、81±7%和75±3%。本研究建立了一种简便灵敏的气相色谱-质谱联用(GC-MS)方法,用于研究异黄酮在大鼠体内的生物利用度、蛋白结合率和代谢情况。结果表明,异黄酮在大鼠体内经静脉和口服给药后,其药代动力学呈线性关系。异黄酮在6.25、12.5和25 mg/kg剂量下的绝对生物利用度分别约为3.85%、33.51%和34.76%。异黄酮素生物利用度低可能是由于其吸收不良或代谢广泛所致。本研究探讨了欧前胡素在大鼠肝微粒体中的I期代谢产物,分离并鉴定了两种代谢产物:黄酮维林和赫拉克林。口服欧前胡素后,在大鼠血浆中检测到一种代谢产物(赫拉克林),在大鼠尿液中检测到两种潜在代谢产物(赫拉克林和黄酮维醇)。然而,在大鼠粪便或胆汁中未检测到任何潜在代谢产物。这些结果表明,欧前胡素代谢产物主要经肾脏排泄,且赫拉克林与活性成分结合。去甲基化和氧化是其主要的代谢途径。体外实验表明,当大鼠血浆中欧前胡素浓度分别为1.0 μg/mL和50.0 μg/mL时,血浆蛋白结合率分别为90.1%和92.6%,表明欧前胡素在细胞内和细胞外空间的分布均相对缓慢。异戊烯基香豆素(IMP)是多种中药的主要成分,具有抗骨质疏松活性。本研究旨在探究IMP的生物转化过程,并评价其代谢产物的抗骨质疏松活性。在筛选的18株丝状真菌中,青霉菌AS 3.510表现出良好的IMP代谢能力,并生成了多种新的衍生物。分离纯化了10种转化产物,并基于光谱数据对其结构进行了精确鉴定。其中8种代谢产物(2-8和10)为新发现,此前未见报道。主要的生物转化反应包括异戊烯基侧链的羟基化和呋喃香豆素骨架的内酯开环。此外,本研究还利用MC3T3-E1细胞评价了所有产物(1-10)的抗骨质疏松活性。结果表明,产物5和8在促进MC3T3-E1细胞生长方面具有最佳的生物活性。这些产物有望用于未来骨质疏松症的治疗。本研究建立了一种简便灵敏的气相色谱-质谱联用方法,用于研究异戊烯酮在大鼠体内的生物利用度、蛋白结合率和代谢情况。结果表明,异戊烯酮在大鼠体内经静脉和口服给药后均呈线性药代动力学特征。异戊烯酮在6.25、12.5和25 mg/kg剂量下的绝对生物利用度分别约为3.85%、33.51%和34.76%。异戊烯酮生物利用度低可能归因于其吸收不良或代谢广泛。本研究还研究了异戊烯酮在大鼠肝微粒体中的I期代谢产物,分离并鉴定了两种代谢产物:黄酮维罗和赫拉克林。口服异戊烯酮后,在大鼠血浆中检测到一种代谢产物(赫拉克林),在大鼠尿液中检测到两种潜在代谢产物(黄酮维罗和赫拉克林)。然而,在大鼠粪便或胆汁中均未检测到潜在的代谢物。这些结果表明,异戊二烯酮代谢物主要经肾脏排泄,且异戊二烯酮与活性成分相关。去甲基化和氧化是其主要的代谢途径。体外实验表明,当大鼠血浆中异戊二烯酮的浓度分别为1.0 μg/mL和50.0 μg/mL时,血浆蛋白结合率分别为90.1%和92.6%,表明异戊二烯酮在细胞内和细胞外空间的分布均相对缓慢。对氧合酶1 (PON1) 是有机磷代谢的关键酶。PON1可通过水解作用使某些有机磷化合物失活。PON1能够水解多种有机磷农药和神经毒剂(如梭曼、沙林和VX)的活性代谢物。 PON1基因多态性的存在导致该酯酶的酶活性水平和催化效率存在差异,提示不同个体可能对有机磷酸酯的毒性作用更为敏感。 吸收:大鼠口服异戊二烯酮吸收缓慢,生物利用度低。然而,由于其他香豆素的协同作用,从提取物(例如,白芷)中吸收迅速。[1] -分布:静脉给药后,血管外分布较少。血浆蛋白结合率超过90%,导致其缓慢分布至细胞内和细胞外间隙。异戊二烯酮易于穿过血脑屏障,且不是P-gp底物。脑内浓度最高的区域为纹状体、海马、脑干、皮层、小脑和间脑。其在海马中的半衰期明显长于其他脑区。 [1] - 代谢:异戊二烯酮在肝微粒体中进行 I 期代谢(去甲基化和氧化),生成黄毒醇和赫拉克林。I 期反应包括单氧化、双氧化和三氧化、内部水解、去甲基化生成羧酸以及 C₅H₈ 基团的丢失。II 期反应包括硫酸盐和葡萄糖醛酸结合。在大鼠尿液中鉴定出 51 种代谢物。[1] - 消除:静脉注射后,异戊二烯酮迅速消除。在大鼠血浆和尿液中检测到黄毒醇和赫拉克林,但在胆汁或粪便中未检测到。[1] - 酶诱导:异戊二烯酮可诱导 I 期(细胞色素 P450)和 II 期(谷胱甘肽 S-转移酶)酶。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
毒性概述
鉴定和用途:欧前胡素是从野芥菜(Imperatorinus arvense)果实中分离得到的呋喃香豆素类化合物。它曾被用作实验药物。人体研究:欧前胡素对人淋巴细胞具有光毒性和光致突变性。体外实验表明,欧前胡素可诱导人淋巴细胞染色体结构畸变和姐妹染色单体交换。动物研究:欧前胡素对小鼠具有抗惊厥作用。用欧前胡素处理的大鼠肝脏未见病变。欧前胡素可诱导中国仓鼠V79细胞和小鼠C3H/1OT1/2细胞中乌本苷基因座的突变。在莱茵衣藻(Chlamydomonas reinhardtii)中,欧前胡素表现出光毒性和光致突变性。异黄酮在Ames试验菌株(TA92、TA97、TA98、TA100)中具有致突变性,但在TA94和TA102菌株中则不具有致突变性。TA98和TA100菌株的致突变性最高。微粒体活化并非致突变性的必要条件。体内和体外研究表明,异黄酮具有抗炎和抗氧化活性。异黄酮是胆碱酯酶(或乙酰胆碱酯酶,AChE)的抑制剂。胆碱酯酶抑制剂(或“抗胆碱酯酶”)抑制乙酰胆碱酯酶的活性。由于乙酰胆碱酯酶在生理过程中发挥着至关重要的作用,因此干扰其活性的化学物质是强效神经毒素;即使是低剂量也会导致唾液分泌过多和流泪,随后出现肌肉痉挛,最终导致死亡。神经毒素和许多农药中的物质已被证实可通过与乙酰胆碱酯酶活性位点的丝氨酸残基结合而发挥作用,从而完全抑制该酶的活性。乙酰胆碱酯酶负责分解神经递质乙酰胆碱,乙酰胆碱在神经肌肉接头处释放,引起肌肉或器官松弛。乙酰胆碱酯酶的抑制会导致乙酰胆碱的积累和持续作用,进而导致持续的神经冲动传递和无法停止的肌肉收缩。最常见的乙酰胆碱酯酶抑制剂是含磷化合物,其设计目的是与该酶的活性位点结合。其结构要求包括一个带有两个亲脂基团的磷原子、一个离去基团(例如卤素或硫氰酸盐)和一个末端氧原子。许多呋喃香豆素的作用机制基于其与DNA和其他细胞成分(如RNA、蛋白质和膜蛋白,包括磷脂酶A2和C、钙依赖性和cAMP依赖性蛋白激酶以及表皮生长因子)形成光加合物的能力。呋喃香豆素可以插入DNA碱基对之间,并在UVA照射下形成环加合物(L579)。 相互作用 本研究在小鼠最大电休克诱导癫痫(MES)模型和烟囱试验中,探讨了异戊二烯酮(IMP)对拉莫三嗪(LTG,一种第二代抗癫痫药物)抗惊厥活性和急性不良反应的影响。为了评估MES试验中IMP和LTG之间的相互作用性质,采用高效液相色谱法(HPLC)测定了脑组织中LTG的总浓度。结果显示,在最大电休克(MES)试验前30分钟腹腔注射50 mg/kg IMP可显著增强拉莫三嗪(LTG)的抗惊厥作用,使其半数有效剂量(ED50)从6.11 mg/kg降至2.47 mg/kg(p < 0.05)。相反,腹腔注射30 mg/kg和40 mg/kg IMP并未显著增强LTG对MES诱发癫痫发作的抗惊厥活性,尽管观察到LTG的ED50值分别从6.11 mg/kg降至5.77 mg/kg和4.28 mg/kg。另一方面,腹腔注射IMP(剂量分别为30、40和50 mg/kg)对LTG的急性不良反应无影响,且在爬杆试验中LTG的半数最大毒性剂量(TD50)几乎保持不变,范围为22.13至30.04 mg/kg。单独使用LTG的保护指数(TD50与ED50的比值)为4.90,而当LTG与IMP(剂量分别为30、40和50 mg/kg)联合使用时,保护指数分别增加至5.21、6.77和8.96。采用高效液相色谱法(HPLC)对颅内总LTG浓度进行药代动力学评价,结果表明,50 mg/kg剂量的IMP不影响实验动物颅内总LTG浓度。因此,在最大电休克试验中观察到的IMP与LTG之间的相互作用本质上是药效学的。本研究表明,在临床前动物研究中,IMP可以改善LTG的药理特性,包括其抗癫痫作用和急性不良反应。如果这些结果能够推广到临床应用,LTG与IMP的联合用药可能对癫痫患者具有重要意义,是一种潜在的有效组合。 背景与目的:草药作为食品和药物被广泛使用,并参与多种生理和病理过程。褪黑激素是由松果体合成和分泌的一种人体激素,具有多种生物学功能。本研究评估了从常见草药中提取的成分对人体褪黑激素代谢的潜在影响。方法:本研究采用体内药代动力学研究(包括12名健康受试者)、体外人肝微粒体(HLM)和重组人细胞色素P450(CYP)同工酶孵育实验,以及基于比较分子场分析和比较分子相似性指数分析的计算机辅助定量构效关系(QSAR)模型分析,以探讨这些相互作用。主要结果:系统筛选66种常用中药后发现,白芷在体外对褪黑素代谢具有最强的抑制作用。体内药代动力学研究表明,白芷可抑制褪黑素代谢,使人体褪黑素的AUC和Cmax分别增加约12倍和4倍。来自白花蛇舌草(Hedyotis diffusa)的香豆素(包括异己烯苷、异己烯苷、叶绿素、5-甲氧基补骨脂素和8-甲氧基补骨脂素)通过抑制人肝微粒体(HLM)中的CYP 1A2、1A1和1B1,显著抑制褪黑素代谢,其Ki值分别为14.5 nM、38.8 nM、6.34 nM、5.34 nM和18 nM。我们建立了一个QSAR模型,该模型能够有效预测香豆素在体内抑制褪黑素代谢的潜在风险。结论和意义:白花蛇舌草中的香豆素在体外和体内均能抑制褪黑素代谢。我们的研究结果为褪黑素的临床应用提供了重要的指导。天然存在的香豆素(NOCs)在小鼠皮肤模型中具有抗癌作用。为了表征NOC化合物对乳腺癌的化学预防潜力,我们首先检测了它们对小鼠乳腺中7,12-二甲基苯并[a]蒽(DMBA)-DNA加合物形成的影响。我们假设,既能抑制细胞色素P450 1A1/1B1又能诱导肝脏谷胱甘肽S-转移酶(GSTs)的NOC化合物,在阻断小鼠乳腺中DMBA-DNA加合物的形成方面最为有效。为了验证这一假设,我们将简单的香豆素类化合物(例如香豆素和柠檬烯,它们能诱导小鼠肝脏GSTs,但对P450 1A1/1B1的影响很小)与线性呋喃香豆素类化合物(例如异异戊二烯酮和异异戊二烯酮,它们能诱导肝脏GSTs,并且是P450 1A1/1B1的强效抑制剂)进行了比较。小鼠预先灌胃给予NOCs(150 mg/kg体重),随后分别接受单次DMBA(50 μg)或多次DMBA(每日20 μg,持续3周和6周)。采用核酸酶P1增强的32P标记法定量乳腺组织中DMBA-DNA加合物的形成。单次DMBA给药后,香豆素、柠檬烯、异异戊二烯酮和异异戊二烯酮分别抑制DMBA-DNA加合物的形成50%、41%、79%和88%。连续给药3周后,香豆素、柠檬烯和异异戊二烯酮分别抑制DMBA-DNA加合物的形成36%、60%和66%;连续给药6周后,它们分别抑制DMBA-DNA加合物的形成0%、49%和55%。在一项为期6周的剂量反应研究中,研究人员测试了选定的NOCs和7,8-苯并黄酮类化合物(一种强效的P4501抑制剂,对GST的影响极小)。结果显示,在35、70和150 mg/kg的剂量下,DMBA-DNA加合物的形成分别被抑制了0%、43%和24%;在异麦芽糖组中,DMBA-DNA加合物的形成分别被抑制了26%、26%和69%;在7,8-苯并黄酮类化合物组中,DMBA-DNA加合物的形成分别被抑制了80%、96%和97%。总之,这些结果表明,与简单的香豆素相比,线性呋喃香豆素对小鼠乳腺中DMBA-DNA加合物的形成具有更强的抑制作用,其主要作用可能是抑制P4501。我们研究了几种先前被鉴定为小鼠肝脏中乙氧卤素-O-脱乙基酶 (EROD) 和/或戊卤素-O-脱烷基酶 (PROD) 强效抑制剂的天然香豆素,以考察它们对小鼠表皮中苯并[a]芘 (B[a]P) 和 7,12-二甲基苯并[a]蒽 (DMBA) DNA 加合物形成的影响,以及它们对这些多环芳烃 (PAHs) 诱导的皮肤肿瘤发展的影响。佛手柑素是一种强效的肝脏 EROD 抑制剂,在首次给予苯并[a]芘 (B[a]P) 前 5 分钟局部应用佛手柑素,并在给药后 24 小时,可显著降低 B[a]P 与 DNA 的总价结合,且呈剂量依赖性。400 nmol 的佛手柑素可使 B[a]P 的共价结合降低 72%。 400 nmol 的佛手柑内酯可显著降低苯并[a]芘 (B[a]P) 的总价结合率达 59%。此外,两种香豆素均选择性地减少了主要 (+) 反式 B[a]P-二醇环氧化物-N2-dGuo 加合物的形成。相反,佛手柑内酯和芫荽苷在 400 nmol 或 800 nmol 的剂量下均未显著降低二甲基苯并咪唑 (DMBA) 与表皮 DNA 的共价结合。异欧前胡素和异欧前胡素是肝脏 PROD 活性的强效抑制剂,在 400 nmol 的剂量下分别显著降低了 DMBA 与表皮 DNA 的总结合率达 67% 和 52%。两种香豆素在相似剂量下也抑制了 B[a]P-DNA 加合物的形成,但程度较轻。 400 nmol 的异异戊二烯酮显著降低了 DMBA 反式和顺式二醇环氧化物衍生的共价 DNA 加合物的形成。佛手柑内酯是 B[a]P 诱导肿瘤发生的强效抑制剂,而芫荽苷在这方面的效果较差。异己香豆素是 DMBA 诱导皮肤肿瘤发生的强效抑制剂,并且可以完全抑制这种多环芳烃的致癌作用。在高于抑制 DMBA 的剂量下,异己香豆素也能抑制 B[a]P 诱导的肿瘤发生。这些发现表明,几种天然存在的香豆素可以阻断多环芳烃(例如苯并[a]芘和二甲基苯并[dMBA])诱导的 DNA 加合物形成和肿瘤起始。它们抑制 DNA 加合物形成和肿瘤起始的机制似乎与抑制参与这些烃代谢活化的 P450 酶有关。最后,某些香豆素对B[a]芘和DMBA诱导的DNA加合物形成和肿瘤起始的不同影响可能有助于阐明特定细胞色素P450酶在其代谢活化中的作用。有关异戊二烯酮(共11种)相互作用的更完整数据,请访问HSDB记录页面。 低细胞毒性:即使在高剂量(5-80 μmol)下,异戊二烯酮在多种细胞系中也显示出低毒性。[1] - 体内安全性:在荷瘤裸鼠中,异戊二烯酮(50-100 mg/kg/天,持续14天)未引起明显的副作用,例如体重减轻、心脏毒性或肝毒性。[1] - 神经毒性:在小鼠爬杆试验中,异戊二烯酮的TD50(神经毒性剂量)范围为329至443 mg/kg。[1] |
| 参考文献 |
|
| 其他信息 |
治疗用途
先前的研究发现,人类日常饮食中会摄入多种天然存在的香豆素。这些香豆素在体外可抑制苯并[a]芘 (B[a]P) 和 7,12-二甲基苯并[a]蒽 (DMBA) 的 P450 介导代谢,阻断小鼠表皮中 DNA 加合物的形成,并且局部应用于小鼠时可抑制 B[a]P 和/或 DMBA 诱导的皮肤肿瘤。本研究旨在探讨两种线性呋喃香豆素化合物(70 mg/kg,连续四天口服)对小鼠不同组织中 P450 和谷胱甘肽 S-转移酶 (GST) 活性以及 B[a]P 和 DMBA 诱导的 DNA 加合物形成的影响。口服异戊二烯酮后1小时和24小时,表皮中乙氧卤素O-脱乙基酶(EROD)和戊基卤素O-脱烷基酶(PROD)的活性显著降低。口服异戊二烯酮1小时后,肺和前胃中EROD的活性略有抑制,而PROD的活性在最后一次口服给药后1小时显著降低。最后一次口服异戊二烯酮24小时后,表皮和肺中EROD和PROD的活性仍然受到抑制。然而,前胃P450的活性已恢复至对照水平。有趣的是,口服异戊二烯酮1小时后,肝脏中EROD的活性受到抑制,但此时PROD的活性未受影响;然而,24小时后,两种酶的活性均升高。EROD和PROD活性的升高与肝脏P450含量的增加相一致。与玉米油对照组相比,口服异戊烯酮和异戊烯酮后1小时和24小时,肝细胞质谷胱甘肽S-转移酶(GST)活性均增加了1.6倍。口服异戊烯酮和异戊烯酮还能抑制苯并[a]芘(B[a]P)和二甲基苯并[a]芘(DMBA)诱导的DNA加合物形成。异戊烯酮预处理可降低前胃中DMBA诱导的DNA加合物形成。异戊烯酮预处理可降低肝脏(B[a]P)、肺(B[a]P)和乳腺上皮细胞(DMBA)中的DNA加合物水平。这些结果表明,膳食补充异戊烯酮和异戊烯酮可能具有潜在的化学预防作用。 /EXPL THER/ 据报道,增强的内皮型一氧化氮合酶 (eNOS) 信号传导与心脏重塑的改善相关,而一氧化氮 (NO) 水平与心脏肥大和心力衰竭相关。异戊烯酮是一种膳食呋喃香豆素,已被证明可以预防自发性高血压大鼠 (SHR) 的心脏肥大。因此,我们旨在阐明异戊烯酮是否通过 NO 信号通路缓解心脏肥大和心力衰竭。在新生小鼠心肌细胞中,异戊烯酮抑制了异丙肾上腺素或苯肾上腺素刺激的蛋白质合成,而 NG-硝基-L-精氨酸甲酯 (L-NAME) 则没有影响。在昆明(KM)雄性小鼠行横向主动脉缩窄(TAC)术后4周,与对照组相比,异戊二烯酮治疗显著降低了心脏重量/体重比(TAC组为6.60 ± 0.35 mg/g,异戊二烯酮15 mg kg⁻¹ d⁻¹灌胃组为4.54 ± 0.29 mg/g,P<0.01);肺重量/体重比(TAC组为7.30 ± 0.85 mg/g,异戊二烯酮15 mg kg⁻¹ d⁻¹灌胃组为5.42 ± 0.51 mg/g)和心肌纤维化程度也观察到类似变化。L-NAME抑制了上述所有改善。异戊二烯酮治疗显著激活了eNOS磷酸化。在TAC小鼠中,利钠肽前体B和NO合酶蛋白抑制剂的心肌mRNA水平升高,而异戊二烯酮治疗可降低这些指标。异戊二烯酮可在体内外减轻心脏肥大,并通过NO介导的通路阻止肥大进展为心力衰竭。本研究采用小鼠最大电休克诱导癫痫(MES)模型和爬杆试验,探讨了异戊二烯酮(IMP)对拉莫三嗪(LTG,一种第二代抗癫痫药物)抗惊厥活性和急性不良反应的影响。为评估MES试验中IMP与LTG的相互作用,采用高效液相色谱法(HPLC)测定了小鼠脑内LTG的总浓度。结果显示,在最大电休克(MES)试验前30分钟腹腔注射50 mg/kg的IMP可显著增强拉莫三嗪(LTG)的抗惊厥作用,使LTG的半数有效剂量(ED50)从6.11 mg/kg降低至2.47 mg/kg(p < 0.05)。相反,腹腔注射30 mg/kg和40 mg/kg的IMP并未显著增强LTG对MES诱发癫痫发作的抗惊厥活性,尽管观察到LTG的ED50值分别从6.11 mg/kg降低至5.77 mg/kg和4.28 mg/kg。另一方面,腹腔注射IMP(剂量分别为30、40和50 mg/kg)对LTG的急性不良反应无影响,烟囱试验中LTG的半数毒性剂量(TD50)几乎保持不变,范围为22.13至30.04 mg/kg。单独使用LTG的保护指数(TD50与ED50的比值)为4.90,而当LTG与IMP(剂量分别为30、40和50 mg/kg)联合使用时,保护指数分别增加至5.21、6.77和8.96。采用高效液相色谱法(HPLC)对颅内总LTG浓度进行药代动力学评价。结果显示,50 mg/kg剂量的IMP不影响实验动物颅内总LTG浓度。因此,在最大电休克试验中观察到的IMP与LTG之间的相互作用本质上是药效学的。本研究表明,在临床前动物研究中,IMP可以改善LTG的药理特性,同时兼顾抗癫痫作用和急性药物不良反应。如果这些结果能够推广到临床应用,LTG与IMP的联合用药可能对癫痫患者具有重要的临床意义,代表一种潜在的优势组合。 /EXPL THER/ 背景:异戊二烯香豆素(IM)是一种从白芷根中分离得到的呋喃香豆素,据报道具有抗惊厥和抗癌作用。本研究检测了IM对九种人类癌细胞系的抗增殖作用,并选择人肝癌HepG2细胞作为IM杀伤的优先靶细胞。此外,本研究还探讨了伊马替尼(IM)诱导细胞凋亡的机制及其在动物体内的作用。方法:采用MTT法检测细胞活力,采用Hoechst染色、Annexin V-PI双染和DNA片段化检测细胞凋亡,采用JC-1染色检测线粒体膜电位,采用Western blot分析检测凋亡相关蛋白的表达。此外,在荷HepG2细胞的裸鼠模型中检测了伊马替尼(IM)的体内抗癌作用。结果:伊马替尼以时间和剂量依赖的方式抑制HepG2细胞增殖,其机制是通过诱导细胞凋亡,具体表现为核形态改变、DNA片段化、磷脂酰丝氨酸外翻、线粒体膜电位丧失、细胞色素c释放入胞质、caspase-3、caspase-8和caspase-9激活以及聚(ADP-核糖)聚合酶(PARP)裂解。由于caspase-8或caspase-9抑制剂可以部分阻止细胞死亡(Western blot分析证实了这一点),我们的结果也表明伊马替尼诱导的细胞凋亡是由死亡受体和线粒体途径共同介导的。在动物模型中,连续14天给予伊马替尼50 mg/kg和100 mg/kg剂量,分别有效抑制肿瘤生长31.93%和63.18%。未观察到明显的体重减轻或宿主毒性。结论:伊马替尼通过死亡受体和线粒体途径诱导HepG2细胞凋亡,从而发挥其抗肿瘤作用。此外,伊马替尼在体内表现出显著的抗肿瘤活性,且体重减轻和宿主损伤可忽略不计。 有关异黄酮类化合物(共9种)治疗用途的更完整数据,请访问HSDB记录页面。 背景:异黄酮(9-[(3-甲基-2-丁烯-1-基)氧基]-7H-呋喃并[3,2-g]色烯-7-酮)是一种呋喃香豆素衍生物,存在于伞形科和芸香科植物中。它在中医中广泛用于治疗多种疾病,包括头痛、牙痛、风湿病,并可用作镇静剂。 [1] - 作用机制:欧前胡素通过增强 GABAA 受体活性、抑制 GABA-T 受体、抑制 AChE/BChE 以及抑制 BACE-1 发挥其中枢神经系统作用。其心血管作用是通过阻断钙通道和激活 eNOS 介导的。其抗癌作用涉及细胞凋亡、自噬和抑制 DNA 加合物形成。[1] - 血脑屏障穿透性:欧前胡素是一种高渗透性化合物,可通过跨细胞被动扩散穿过血脑屏障,且不是 P-糖蛋白的底物。[1] - 治疗潜力:欧前胡素在癫痫、焦虑症、抑郁症、阿尔茨海默病、帕金森病、心血管疾病(高血压、心肌肥大)、癌症、骨质疏松症、细菌感染、HIV 和炎症性疾病方面具有潜在的应用价值。[1] |
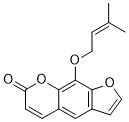
| 分子式 |
C16H14O4
|
|---|---|
| 分子量 |
270.284
|
| 精确质量 |
270.089
|
| CAS号 |
482-44-0
|
| PubChem CID |
10212
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
448.3±45.0 °C at 760 mmHg
|
| 熔点 |
98-100ºC
|
| 闪点 |
224.9±28.7 °C
|
| 蒸汽压 |
0.0±1.1 mmHg at 25°C
|
| 折射率 |
1.606
|
| LogP |
3.81
|
| tPSA |
52.58
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
20
|
| 分子复杂度/Complexity |
436
|
| 定义原子立体中心数目 |
0
|
| SMILES |
O(C([H])([H])/C(/[H])=C(\C([H])([H])[H])/C([H])([H])[H])C1=C2C(C([H])=C([H])C(=O)O2)=C([H])C2C([H])=C([H])OC1=2
|
| InChi Key |
OLOOJGVNMBJLLR-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C16H14O4/c1-10(2)5-7-19-16-14-12(6-8-18-14)9-11-3-4-13(17)20-15(11)16/h3-6,8-9H,7H2,1-2H3
|
| 化学名 |
9-(3-methylbut-2-enoxy)furo[3,2-g]chromen-7-one
|
| 别名 |
Ammidin MarmelosinImperatorin Pentosalen
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~50 mg/mL (~184.99 mM)
|
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (9.25 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (9.25 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL 澄清 DMSO 储备液加入到 900 μL 玉米油中并混合均匀。 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.6999 mL | 18.4993 mL | 36.9987 mL | |
| 5 mM | 0.7400 mL | 3.6999 mL | 7.3997 mL | |
| 10 mM | 0.3700 mL | 1.8499 mL | 3.6999 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
|
|
|
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1


































 463611831
463611831
