| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|
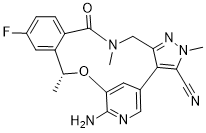
洛拉替尼(以前也称为洛拉替尼,PF-06463922;商品名:洛布雷纳)是一种强效、口服生物利用度高、可穿透血脑屏障的ATP竞争性ALK/ROS1双重抑制剂,具有潜在的抗肿瘤活性。 PF-06463922 的 Ki 值分别小于 0.02 nM、0.07 nM 和 0.7 nM,可抑制 ROS1、ALK(野生型)和 ALK(L1196M)。FDA 已批准洛拉替尼用于治疗间变性淋巴瘤激酶 (ALK) 阳性的转移性非小细胞肺癌患者。PF-06463922 给药后,可与 ROS1 激酶和 ALK 激酶结合并抑制其活性。这会破坏 ALK 和 ROS1 介导的信号通路,最终抑制肿瘤细胞生长。除了治疗 ROS1 融合阳性癌症(包括需要具有中枢神经系统穿透能力的药物的癌症)外,PF-06463922 还可能逆转由 ROS1 突变引起的克唑替尼耐药性。
| 靶点 |
ALKL1196 (IC50 = 15-43 nM); ALKG1269A (IC50 = 14-80 nM); ALK1151Tins (IC50 = 38-50 nM); ALKG1202R (IC50 = 77-113 nM); ALKWT (IC50 <0.07 nM); ALKL1996M (IC50 = 0.6 nM); ALKG1269A (IC50 = 0.9 nM); ALK1151Tins (IC50 = 0.1 nM); ALKL1152R (IC50 <0.1 nM); ALKS1206Y (IC50 = 0.2 nM); ALKC1156Y (IC50 <0.1 nM); ALKF1174L (IC50 <0.1nM)
Anaplastic lymphoma kinase (ALK) (wild-type Ki <0.07 nM; L1196M mutant Ki 0.7 nM) c-ros oncogene 1 (ROS1) (Ki <0.02 nM) [1] |
|---|---|
| 体外研究 (In Vitro) |
PF-06463922 对多种 ALK 临床突变均表现出抑制活性,IC50 值介于 0.2 至 77 nM 之间,并且对 ALK 具有显著的细胞活性。[1] 在携带 SLC34A2-ROS1 融合基因的 HCC78 人非小细胞肺癌细胞和表达人 CD74-ROS1 的 BaF3-CD74-ROS1 细胞中,PF-06463922 可显著降低细胞增殖并诱导细胞凋亡。 [2] 在表达非突变 ALK 或突变 ALK 融合蛋白的 NSCLC 细胞中,PF-06463922 也表现出强烈的生长抑制活性并引起细胞凋亡。[3]在用 EML4-ALK 改造的 NIH-3T3 细胞中,PF-06463922 抑制野生型 ALK 磷酸化,IC50 为 1.3 nM。针对临床报道的ALK突变体,该化合物显示出强效的细胞活性:F1174L IC50 = 0.2 nM,C1156Y IC50 = 1.6 nM,G1269A IC50 = 1 nM,S1206Y IC50 = 5 nM,L1196M IC50 = 4.2 nM,L1152R IC50 = 2 nM,以及G1202R IC50 = 77 nM,与克唑替尼(化合物1)相比,其效力提高了40至825倍。[1]
在针对206种重组激酶的生化激酶筛选中,PF-06463922对大多数脱靶激酶表现出超过100倍的选择性;最接近的脱靶突变体为:LTK (TYK1) Ki = 2.7 nM(与 ALK-L1196M 相比为 3.9 倍),FER Ki = 3.3 nM(4.7 倍),FES (FPS) Ki = 6.0 nM(8.6 倍),PTK2B (FAK2) Ki = 14 nM(20 倍),TNK2 (ACK) Ki = 17 nM(24 倍),PTK2 (FAK) Ki = 17 nM(24 倍),NTRK2 (TRKB) Ki = 23 nM(33 倍),NTRK1 (TRKA) Ki = 24 nM(34 倍),NTRK3 (TRKC) Ki = 46 nM(66 倍),以及 FRK (PTK5) Ki = 53 nM(76 倍)。[1] |
| 体内研究 (In Vivo) |
PF-06463922 具有 P-糖蛋白 1 介导的外排倾向低、分布容积适中、半衰期合理、血浆清除率低以及在大鼠体内生物利用度为 100% 的特点。[1] PF-06463922 在表达人 CD74-ROS1 和 Fig-ROS1 的 NIH3T3 异种移植模型中表现出细胞减灭抗肿瘤功效,其作用机制是通过抑制下游信号分子和 ROS1 磷酸化,以及抑制肿瘤细胞周期蛋白 Cyclin D1。[2]在表达 EML4-ALK、EML4-ALK-L1196M、EML4-ALK-G1269A、EML4-ALK-G1202R 或 NPM-ALK 的肿瘤异种移植小鼠中,PF-06463922 也表现出强大的体内抗肿瘤活性。[3]
|
| 酶活实验 |
微流控迁移率分析用于测定重组人野生型和突变型ALK激酶结构域蛋白(氨基酸1093-1411)的激酶活性。这些蛋白由本实验室通过杆状病毒表达和MgATP自磷酸化制备。反应体系包含3 μM 5-FAM-KKSRGDYMTMQIG-CONH2、5 mM MgCl2、1.3 nM野生型ALK或0.5 nM突变型ALK(反应1小时后肽底物磷酸化率可达15-20%),以及在25 mM Hepes缓冲液(pH 7.1)中测定的ATP Km值。动力学和晶体学研究结果表明,这些抑制剂是ATP竞争性的。将转化率(%)拟合到竞争性抑制方程即可得到Ki值。 ROS1酶的检测方法与ALK相同,不同之处在于使用0.25 nM重组人ROS1催化结构域(氨基酸1883-2347)。采用206种激酶组成的激酶库来评估激酶抑制剂的选择性。
重组人野生型和突变型ALK激酶结构域蛋白(氨基酸1093-1411)通过杆状病毒表达产生,经MgATP自磷酸化预激活后,采用微流控迁移率变动分析法检测其激酶活性。反应体系包含 1.3 nM 野生型 ALK 或 0.5 nM 突变型 ALK(1 小时后肽底物磷酸化率达到 15-20%)、3 μM FAM 标记的肽底物 (5-FAM-KKSRGDYMTMQIG-CONH2)、5 mM MgCl2 和 Km 水平的 ATP,溶于 25 mM Hepes 缓冲液 (pH 7.1) 中。动力学和晶体学研究表明,抑制剂与 ATP 竞争。Ki 值通过将转化率 (%) 拟合到竞争性抑制方程来计算。[1] ROS1 酶的活性测定方法类似,使用 0.25 nM 重组人 ROS1 催化结构域(氨基酸 1883-2347)。 [1] 使用包含 206 种激酶的激酶库,通过基于荧光共振能量转移 (FRET) 的 Z'-LYTE 检测(偶联酶形式,磷酸化肽与非磷酸化肽的差异性蛋白水解切割)、基于时间分辨 FRET (TR-FRET) 的 Adapta 检测(使用 Alexa Fluor 647 标记的 ADP 示踪剂和 Eu 标记的抗 ADP 抗体)或基于 TR-FRET 的 LanthaScreen 结合检测(使用 Alexa Fluor 示踪剂和 Eu 标记的抗标签抗体)评估激酶选择性。ATP 浓度接近表观 Km 值。[1] |
| 细胞实验 |
在96孔板中,将细胞接种于含10% FBS的生长培养基中,并在37°C下孵育过夜。第二天,向指定孔中加入不同浓度的洛拉替尼或合适的对照品,并在37°C下继续孵育72小时。为了确定细胞的相对数量,进行CellTiter-Glo检测。采用四参数分析方法拟合浓度-反应曲线并确定IC50值。
将细胞以20,000个/孔的密度接种于96孔板中,并在含0.5%血清的生长培养基中过夜孵育。PF-06463922用无血清培养基稀释后加入细胞中,孵育1小时,然后用真空抽吸法去除。制备细胞裂解液,并使用化学发光夹心ELISA试剂盒测定磷酸化ALK(Tyr1604)水平。使用另一套夹心ELISA试剂盒测定总ALK水平。采用四参数分析法,通过浓度-反应曲线拟合计算IC50值。[1] |
| 动物实验 |
如前所述,在LSL-FIG-ROS1;Cdkn2a−/−;LSL-Luc小鼠中,通过颅内立体定向注射Adeno-Cre诱导新生胶质母细胞瘤(GBM)的发生。生物发光成像(BLI)用于追踪肿瘤的发展,具体方法将在下文讨论。当肿瘤达到特定大小(107 p-1·s-1·cm-2·s-1)时,将动物随机分配至载体对照组或接受为期3天、7天或14天的lerlatinib治疗组,治疗剂量按规定剂量进行。药物通过皮下植入的Alzet渗透泵给药。治疗结束后,对GBM肿瘤进行显微解剖,将组织快速冷冻于液氮中,然后处死小鼠。剩余的脑组织用于组织学分析。
大鼠药代动力学研究:雄性大鼠(具体情况未说明)单次静脉注射1 mg/kg或单次口服10 mg/kg的PF-06463922。两种给药途径的制剂均为乙醇/PEG200/水(10:40:50)溶液。在不同时间点采集血样。为评估中枢神经系统生物利用度,大鼠单次口服10 mg/kg,并在给药后1、4、7、12和24小时采集脑脊液(CSF)、脑组织和血浆样本。分析脑组织和血浆样本中的药物浓度,并使用游离药物分数计算游离AUC。[1] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
单次口服100 mg劳拉替尼后,达峰时间(Tmax)中位数为1.2小时(0.5至4小时);每日一次口服100 mg达到稳态后,达峰时间中位数为2小时(0.5至23小时)。与静脉给药相比,口服给药后的平均绝对生物利用度为81%(90% CI 75.7%, 86.2%)。劳拉替尼与高脂肪、高热量餐(约1000卡路里,其中150卡路里来自蛋白质,250卡路里来自碳水化合物,500至600卡路里来自脂肪)同时服用,对劳拉替尼的药代动力学无临床意义上的影响。单次口服100 mg放射性标记的劳拉替尼后,48%的放射性物质从尿液中回收(<1%为原形),41%从粪便中回收(约9%为原形)。 单次静脉给药后,平均(CV%)稳态分布容积(Vss)为305 L(28%)。 单次口服100 mg后,平均口服清除率(CL/F)为11 L/h(35%),稳态时增加至18 L/h(39%),提示存在自身诱导作用。 代谢/代谢物体外研究表明,劳拉替尼主要通过CYP3A4和UGT1A4代谢,CYP2C8、CYP2C19、CYP3A5和UGT1A3的代谢活性较低。在血浆中,由劳拉替尼酰胺键和芳香醚键氧化裂解产生的苯甲酸代谢物 (M8) 占人体 [14C] 质量平衡研究中循环放射性的 21%。这种氧化裂解代谢物 M8 没有药理活性。 生物半衰期 单次口服 100 mg 劳拉替尼后,平均血浆半衰期 (t½) 为 24 小时 (40%)。 PF-06463922 表现出较低的血浆清除率 (CLp = 15.5 mL/min/kg,静脉注射 1 mg/kg 后)、中等的分布容积 (Vav = 2.7 L/kg) 和合理的半衰期 (t1/2 = 2.7 小时)。口服10 mg/kg剂量后,Cmax = 1727 ng/mL,Tmax = 1.3 h,AUC0-24 = 14933 ng·h/mL,绝对口服生物利用度 (F) = 100%。[1] 体外人肝微粒体固有清除率 (Clint,app) <8 mL/min/kg。[1] MDCK-MDR1 BA/AB 外排比为 1.5(底物浓度为 2 μM,pH 7.4),表明 P-糖蛋白介导的外排作用较弱。 [1] 大鼠中枢神经系统渗透性:单次口服 10 mg/kg 剂量后,脑脊液 AUC = 3475 nM·h,游离脑组织 AUC = 1235 nM·h(脑组织 fu = 0.12),游离血浆 AUC = 11052 nM·h(血浆 fu = 0.36)。AUC 比值分别为:脑脊液/游离血浆 = 0.31,游离脑组织/游离血浆 = 0.21。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
在早期的大型临床试验中,接受标准剂量劳拉替尼治疗的患者中,高达 28% 出现血清转氨酶水平升高,但仅有 2% 的患者转氨酶水平超过正常值上限的 5 倍,很少导致提前停药。这些异常通常是短暂的、无症状的,且不伴有黄疸。上市前临床试验中未观察到具有临床意义的肝损伤病例。相比之下,大多数其他 ALK 抑制剂与急性肝损伤病例相关,这些肝损伤可能很严重,甚至致命。肝损伤通常发生在开始治疗后的 4 至 12 周内,表现为血清转氨酶水平显著升高,随后出现黄疸和进行性肝功能障碍。虽然尚未发现劳拉替尼与严重肝损伤相关,但其临床应用受到限制。概率评分:E(未经证实,但怀疑是导致具有临床意义的肝损伤的罕见原因)。 妊娠和哺乳期影响 ◉ 哺乳期用药概述 目前尚无关于劳拉替尼在哺乳期临床应用的信息。生产商建议在劳拉替尼治疗期间以及末次给药后7天内停止母乳喂养。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对泌乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白结合 体外研究表明,劳拉替尼在2.4 µM浓度下与血浆蛋白的结合率为66%。血血浆比为0.99。 大鼠血浆蛋白结合率:游离分数 (fu) = 0.36(在2.4 μM浓度下测定)。大鼠脑组织结合:未结合分数(fu)= 0.12。[1] |
| 参考文献 | |
| 其他信息 |
药效学
基于B7461001研究的数据,在推荐剂量下达到稳态暴露时,观察到3级或4级高胆固醇血症以及任何3级或4级不良事件的暴露-反应关系。不良事件的发生概率随劳拉替尼暴露量的增加而增加。在同一项B7461001研究中,295例接受每日一次100 mg劳拉替尼推荐剂量并进行心电图监测的患者中,PR间期较基线的最大平均变化为16.4 ms(双侧90%置信区间[CI]上限为19.4 ms)。在284例基线PR间期<200 ms的患者中,14%的患者在开始服用劳拉替尼后出现PR间期延长≥200 ms。PR间期延长与药物浓度相关,1%的患者出现房室传导阻滞。最后,在B7461001研究的活性评估部分,在接受推荐剂量劳拉替尼治疗的275例患者中,未检测到QTcF间期显著延长(即>20 ms)。 PF-06463922(也称为劳拉替尼、洛布雷纳,CAS号1454846-35-5)是由辉瑞公司发现的一种大环ALK/ROS1抑制剂。它采用大环酰胺模板设计,以实现高亲脂效率(LipE)、低分子量和最佳的log D值(2.3),从而实现中枢神经系统穿透。该化合物目前正在进行I/II期临床试验(NCT01970865),用于治疗ALK阳性非小细胞肺癌,特别是脑转移或耐药突变患者。[1] |
| 分子式 |
C21H19FN6O2
|
|---|---|
| 分子量 |
406.41
|
| 精确质量 |
406.155
|
| 元素分析 |
C, 62.06; H, 4.71; F, 4.67; N, 20.68; O, 7.87
|
| CAS号 |
1454846-35-5
|
| 相关CAS号 |
1924207-18-0 (acetate);2135926-03-1;1454846-35-5;2306217-6 (hydrate);
|
| PubChem CID |
71731823
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.4±0.1 g/cm3
|
| 沸点 |
675.0±55.0 °C at 760 mmHg
|
| 闪点 |
362.1±31.5 °C
|
| 蒸汽压 |
0.0±2.1 mmHg at 25°C
|
| 折射率 |
1.687
|
| LogP |
1.24
|
| tPSA |
110.06
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
7
|
| 可旋转键数目(RBC) |
0
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
700
|
| 定义原子立体中心数目 |
1
|
| SMILES |
FC1C([H])=C([H])C2C(N(C([H])([H])[H])C([H])([H])C3C(=C(C#N)N(C([H])([H])[H])N=3)C3C([H])=NC(=C(C=3[H])O[C@]([H])(C([H])([H])[H])C=2C=1[H])N([H])[H])=O
|
| InChi Key |
IIXWYSCJSQVBQM-LLVKDONJSA-N
|
| InChi Code |
InChI=1S/C21H19FN6O2/c1-11-15-7-13(22)4-5-14(15)21(29)27(2)10-16-19(17(8-23)28(3)26-16)12-6-18(30-11)20(24)25-9-12/h4-7,9,11H,10H2,1-3H3,(H2,24,25)/t11-/m1/s1
|
| 化学名 |
(16R)-19-amino-13-fluoro-4,8,16-trimethyl-9-oxo-17-oxa-4,5,8,20-tetrazatetracyclo[16.3.1.02,6.010,15]docosa-1(22),2,5,10(15),11,13,18,20-octaene-3-carbonitrile
|
| 别名 |
Lorbrena; PF06463922; PF-6463922; PF6463922; PF 6463922; PF 06463922; PF-06463922;Lorlatinib; 1454846-35-5; Lorbrena; Lorviqua; lorlatinibum; PF06463922
|
| HS Tariff Code |
2934.99.09.01
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~81 mg/mL (~199.3 mM)
Water: <1 mg/mL Ethanol: ~30 mg/mL warmed (~73.8 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (6.15 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: 2.5 mg/mL (6.15 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 悬浊液; 超声助溶。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (6.15 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: ≥ 2.5 mg/mL (6.15 mM) (饱和度未知) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 配方 5 中的溶解度: 2% DMSO+30% PEG 300+ddH2O: 5mg/mL 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4606 mL | 12.3028 mL | 24.6057 mL | |
| 5 mM | 0.4921 mL | 2.4606 mL | 4.9211 mL | |
| 10 mM | 0.2461 mL | 1.2303 mL | 2.4606 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Analysis of the Effectiveness and Safety of Lorlatinib in Untreated ALK-Positive NSCLC Patients in a French Real-World Context
CTID: NCT06487078
Phase: N/A Status: Not yet recruiting
Date: 2024-11-08
PF-06463922 is a potent inhibitor of ROS1.Proc Natl Acad Sci U S A. 2015 Mar 17; 112(11): 3493–3498. |
PF-06463922 inhibits crizotinib-induced ROS1 mutants.Proc Natl Acad Sci U S A. 2015 Mar 17; 112(11): 3493–3498. |
(A) Comparison of ROS1 crystal structures bound with PF-06463922 (green) and crizotinib (magenta). (B) PF-06463922 interactions with ROS1 and the PF-06463922 ROS1 binding site.Proc Natl Acad Sci U S A. 2015 Mar 17; 112(11): 3493–3498. |
PF-06463922 inhibits ROS1 fusion-driven tumorigenesis in vivo.Proc Natl Acad Sci U S A. 2015 Mar 17; 112(11): 3493–3498. |
PF-06463922 inhibits FIG-ROS1–mediated tumor growth in a model of GBM. (A) Representative photomicrographs of bioluminescent imaging of a mouse genetically engineered to develop a GBM and its response to a 7- and 14-d treatment with PF-06463922. (B) Decrease in BLI output 7 and 14 d post treatment.Proc Natl Acad Sci U S A. 2015 Mar 17; 112(11): 3493–3498. |
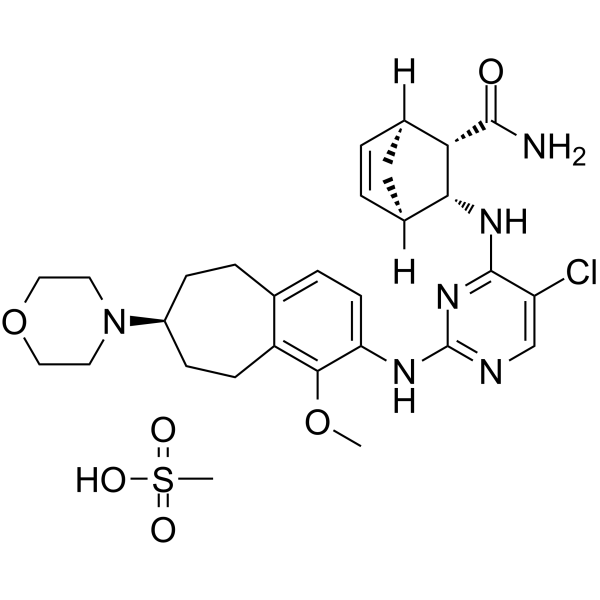 CEP-28122 mesylate salt
CEP-28122 mesylate salt
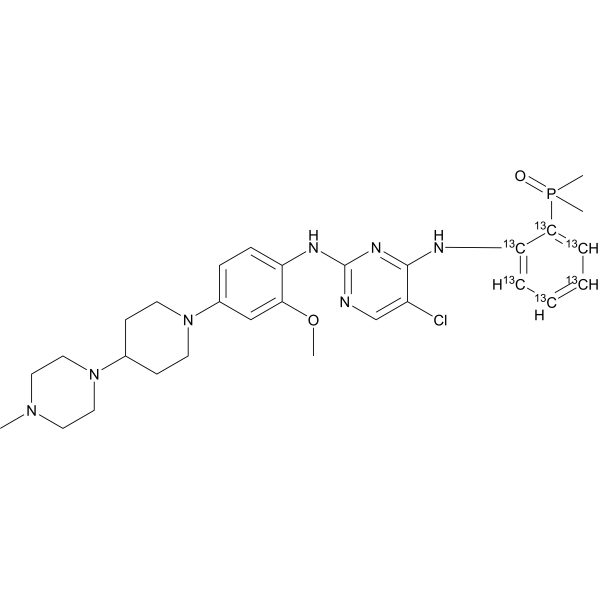 Brigatinib-13C6 (AP-26113-13C6)
Brigatinib-13C6 (AP-26113-13C6)
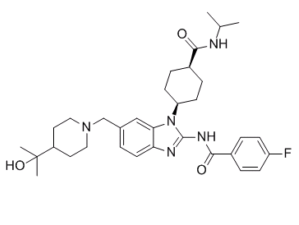 贝扎替尼
贝扎替尼
 双盐酸盐色瑞替尼
双盐酸盐色瑞替尼
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
")
")
")
")
")
 463611831
463611831
