| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|
| 靶点 |
PDE4
S-(+)-Rolipram (ME-3167; SB95952; ZK-62711) selectively inhibits cyclic 3',5'-monophosphate phosphodiesterase type IV (PDE4, also known as type IV phosphodiesterase). Confirmed that its inhibitory activity against PDE4 is significantly stronger than that of its optical isomer R-(-)-Rolipram [2] |
|---|---|
| 体外研究 (In Vitro) |
(+)-Rolipram(0.015-1000 μM;20 小时)以剂量依赖性方式抑制 LPS 产生的人单核细胞 (MNC) TNF 的 IC50 为 550 nM[1]。
|
| 体内研究 (In Vivo) |
(+)-咯利普兰剂量依赖性地抑制大鼠的运动活动并引起头部抽搐(0.025–6.25 mg/kg;腹膜内注射)[2]。当腹腔注射 0.06–25 mg/kg (+)-咯利普兰时,大鼠的直肠温度随剂量成比例下降[2]。
在食物驱动的双杠杆操作任务中,训练Long Evans大鼠区分0.2mg/kg IP(+/-)-rolipram和赋形剂注射。九只大鼠中有八只在平均91次训练后获得了辨别力(最小65次,最大137次)。(+/-)-罗利普兰的ED50为0.06mg/kg IP。对(-)-和(+)-罗利普兰的泛化测试表明,(-)异构体的活性是(+)罗利普兰的8倍,ED50分别为0.06和0.4 mg/kg IP。磷酸二酯酶抑制剂RO 20-1724在0.6和1.0 mg/kg IP的剂量下部分(83%)推广到(+/-)-罗利普兰。IBMX 5mg/kg IP显示63%的泛化率。对丙咪嗪和去甲肾上腺素摄取抑制剂奥沙普林的(+)-和(-)-异构体的测试表明,NA摄取抑制药物不会形成(+/-)-罗利普兰样的感受内线索。dbcAMP 12.5mg/kg SC和100mg/kg SC dbcGMP不能推广到训练药物。这种剂量的(+/-)-罗利普兰在大鼠体内产生的鉴别刺激的性质仍有待阐明[3]。 1. 在雄性Wistar大鼠体内,S-(+)-罗利普兰给药后表现出剂量依赖性的神经活性:当剂量为0.3 mg/kg(腹腔注射,ip)时,可显著增加大鼠的自发运动活性(通过活动监测仪记录)——与对照组(给予含少量助溶剂的生理盐水的大鼠)相比,5分钟内的水平移动距离和站立次数分别增加约30%和40%;当剂量为3 mg/kg(ip)时,除运动活性增强外,大鼠直肠温度出现轻度下降,较基线降低0.5-1.0℃;当剂量升高至10 mg/kg(ip)时,大鼠出现明显共济失调:在旋转杆实验(转速10 rpm)中,大鼠在旋转杆上的停留时间显著缩短至90秒以内(最大观察时间为180秒),较对照组减少50%以上[2] 2. 与罗利普兰的另一光学异构体R-(-)-罗利普兰相比,相同剂量下的S-(+)-罗利普兰对运动活性增强和体温调节的作用更显著。例如,在0.3 mg/kg(ip)剂量下,S-(+)-罗利普兰诱导的自发运动活性增加幅度是R-(-)-罗利普兰的2倍;此外,S-(+)-罗利普兰起效更快——给药后15-30分钟达到效应峰值(如最大运动活性或体温变化),而R-(-)-罗利普兰需30-60分钟才能达到效应峰值[2] |
| 酶活实验 |
用罗利普兰抑制PDE4以表皮生长因子受体依赖的方式促进P2Y11/IL-1R诱导的CXCR7表达和CCL20产生的上调。使用天然表达CXCR7但缺乏CXCR4的星形细胞瘤细胞系,P2Y11/IL-1R激活有效地诱导了CCL20的产生,即使在没有PDE4抑制的情况下,CXCR7激动剂TC14012也能增强CCL20的生产。此外,RNA干扰导致的CXCR7耗竭抑制了CCL20的产生。在巨噬细胞中,P2Y11和CXCR7被其各自的激动剂同时激活足以诱导CCL20的产生,而不需要PDE4的抑制,因为CXCR7的激活增加了其自身并消除了CXCR4的表达。最后,对巨噬细胞分泌组中多种CCL趋化因子的分析表明,CXCR4失活和CXCR7活化选择性地增强了P2Y11/IL-1R介导的CCL20分泌。总之,我们的数据确定CXCR7是P2Y11/IL-1R启动的信号级联的组成部分,CXCR4相关的PDE4是调节检查点。Cell Mol Life Sci. 2024 Mar 13;81(1):132.
|
| 细胞实验 |
抑制肿瘤坏死因子α产生的化合物在感染性休克动物模型中具有保护作用。最近的研究表明,黄嘌呤衍生物具有有益的作用,它通过充当非特异性cAMP磷酸二酯酶抑制剂来抑制肿瘤坏死因子α的产生。在这个实验中,我们测试了(+/-)-罗利普兰(外消旋体)及其对映体对脂多糖(LPS)刺激的人单核细胞的影响。罗利普兰具有苯基吡咯烷酮结构,与甲基黄嘌呤无关,是IV型磷酸二酯酶的特异性抑制剂。我们的研究结果表明,罗利普兰是LPS诱导的肿瘤坏死因子α合成的一种非常有效的抑制剂。与非特异性抑制剂己酮可可碱相比,(+/-)-罗利普兰(130 nM)的IC50低500多倍。罗利普兰对肿瘤坏死因子α产生的影响取决于分子的空间构型,因为(-)-对映体的IC50比(+)-对异构体低五倍。所有受试物质的抑制作用对肿瘤坏死因子α而非白细胞介素-1β具有选择性,因为白细胞介蛋白-1β的产生仅受到轻微影响[1]。
|
| 动物实验 |
溶于100%聚乙二醇(PEG)中,配制成适当浓度;1 mL/kg;静脉注射。
雄性哈特利豚鼠。本研究旨在探讨磷酸二酯酶抑制剂罗利普兰对脑组织再生的影响。通过单次腹腔注射氯化三甲基锡(2.2 mg/kg),构建了三甲基锡注射小鼠模型,该模型可用于研究海马组织再生。从注射三甲基锡的第二天开始,每天腹腔注射罗利普兰(10 mg/kg),直至取样前一天。在实验1中,于注射三甲基锡后第7天进行强迫游泳试验,并采集脑组织样本。在实验2中,于注射三甲基锡后第3-5天腹腔注射溴脱氧尿苷(150 mg/kg/天),并在注射三甲基锡后第21天进行取样。样本常规进行石蜡包埋,并切片用于组织病理学研究。在实验1中,罗利普兰治疗组小鼠在强迫游泳实验中表现出更短的静止不动时间。组织病理学分析显示,罗利普兰治疗促进了齿状回中神经元核阳性神经元的补充,同时伴有磷酸化环磷酸腺苷反应元件结合蛋白(pCREB)阳性细胞比例的增加。此外,罗利普兰还降低了具有活化形态的离子化钙结合衔接蛋白1(ICB1)阳性小胶质细胞的比例以及肿瘤坏死因子-α(TNF-α)表达细胞的数量。在实验2中,溴脱氧尿苷/神经元核双重免疫荧光染色显示,罗利普兰治疗组小鼠中双阳性细胞数量增加。这些结果表明,罗利普兰通过增强新生神经元的存活率和抑制神经炎症,有效促进脑组织再生。Neuroreport. 2024年9月4日;35(13):832-838。 1. 动物选择和饲养:使用体重200-250克的雄性Wistar大鼠。实验前,大鼠在饲养环境中适应1周,温度为22±2℃,相对湿度为50±5%,光照/黑暗周期为12小时(7:00至19:00开灯)。大鼠可自由摄取标准实验室饲料和自来水[2]。 2. 药物制备:将S-(+)-罗利普兰溶于生理盐水中,并加入少量增溶剂(浓度<5%,以确保完全溶解)。配制浓度分别为0.03 mg/mL、0.3 mg/mL、1 mg/mL和3 mg/mL的储备液。给药体积根据大鼠体重计算,每100 g体重给予0.1 mL药物溶液[2] 3. 给药途径和剂量组:采用腹腔注射(ip)途径给药。设置四个剂量组:0.03 mg/kg、0.3 mg/kg、3 mg/kg和10 mg/kg,每组6只大鼠。对照组给予等体积的生理盐水,其中含有相同量的增溶剂[2] 4. 检测指标和时间点:① 自发运动活性:给药后15、30、60和120分钟,将每只大鼠放入活动监测箱(25 cm×25 cm×30 cm)中,测量5分钟内的水平移动距离(由红外传感器记录)和直立行为次数(由垂直传感器记录)。 ② 共济失调:给药后30分钟,将大鼠置于转速为10 rpm的转棒上,记录首次跌倒的时间(最长观察时间:180秒)。③ 体温:给药前以及给药后30分钟和60分钟,使用直肠温度计测量直肠温度(将温度计插入直肠2厘米,保持30秒以稳定读数)[2] |
| 毒性/毒理 (Toxicokinetics/TK) |
在文献[2]报道的0.03-10 mg/kg(腹腔注射)的实验剂量范围内,S-(+)-罗利普兰未引起大鼠死亡或严重的组织损伤。然而,在10 mg/kg的高剂量下,它可诱发大鼠共济失调,持续约60分钟——症状包括行走不稳和肢体协调性下降,这些症状在给药后120分钟自行缓解。此外,3-10 mg/kg剂量组的大鼠表现出短暂的摄食量下降:给药后60分钟内,摄食量较对照组减少了20-30%。未观察到其他明显的毒性反应(如呕吐、腹泻或毛发蓬乱)。[2]
|
| 参考文献 |
|
| 其他信息 |
抑制肿瘤坏死因子-α (TNF-α) 生成的化合物在脓毒性休克动物模型中具有保护作用。近期研究表明,黄嘌呤衍生物具有有益作用,它们通过作为非特异性 cAMP 磷酸二酯酶抑制剂来抑制 TNF-α 的生成。在本实验中,我们测试了 (+/-)-罗利普兰(外消旋体)及其对映体对脂多糖 (LPS) 刺激的人单核细胞的影响。罗利普兰具有苯基吡咯烷酮结构,与甲基黄嘌呤类化合物无关,并且是 IV 型磷酸二酯酶的特异性抑制剂。我们的结果表明,罗利普兰是 LPS 诱导的 TNF-α 合成的强效抑制剂。与非特异性抑制剂己酮可可碱相比,(+/-)-罗利普兰的 IC50 值 (130 nM) 低 500 倍以上。罗利普兰对肿瘤坏死因子-α生成的影响取决于分子的空间构型,因为(-)-对映异构体的IC50值比(+)-对映异构体低五倍。所有受试物质的抑制作用均选择性地针对肿瘤坏死因子-α而非白细胞介素-1β,因为白细胞介素-1β的生成仅受到轻微影响。[1]
已研究了选择性环磷酸腺苷(cAMP)磷酸二酯酶(PDE)抑制剂(+/-)-罗利普兰及其光学异构体(0.006至25 mg kg-1)在诱导大鼠出现特征性行为改变(如体温过低、活动减少、前爪抖动、梳理毛发和头部抽搐)方面的功效。 (+)-罗利普兰的效力比外消旋体低约15倍,提示其与大鼠脑内cAMP磷酸二酯酶同工酶存在立体选择性相互作用。脑内给药后,立体异构体也表现出异常的效力比。这些发现表明,(+)-罗利普兰在体内作为神经营养性PDE抑制剂的效力低于其(-)-对映体。[2] 1. S-(+)-罗利普兰是罗利普兰的药理活性光学异构体,罗利普兰是一种选择性PDE4抑制剂。其神经营养作用主要通过抑制PDE4催化的细胞内环磷酸腺苷(cAMP)水解来实现,从而提高细胞内cAMP水平。 cAMP 升高进一步调节神经递质释放(例如多巴胺、去甲肾上腺素)和细胞内信号通路(例如 PKA 信号通路),最终导致运动活动、体温和运动协调性的变化[2] 2. 文献[2]中的研究目的是比较罗利普兰的两种光学异构体(S-(+)-和 R-(-)-罗利普兰)在大鼠体内的神经营养作用。结果证实,S-(+)-罗利普兰是罗利普兰发挥神经活性作用(如运动活性调节和体温调节)的主要异构体,为开发光学异构体特异性PDE4抑制剂和进一步研究罗利普兰的作用机制提供了实验证据[2]。3. 文献[1]和[3]仅研究了外消旋罗利普兰(S-(+)-和R-(-)-异构体的混合物)的作用,并未具体提及S-(+)-罗利普兰;因此,这两篇文献[1][3]中未提取到关于S-(+)-罗利普兰的相关信息。 |
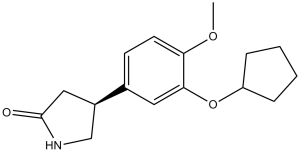
| 分子式 |
C16H21NO3
|
|
|---|---|---|
| 分子量 |
275.34
|
|
| 精确质量 |
275.152
|
|
| 元素分析 |
C, 69.79; H, 7.69; N, 5.09; O, 17.43
|
|
| CAS号 |
85416-73-5
|
|
| 相关CAS号 |
Rolipram;61413-54-5;(R)-(-)-Rolipram;85416-75-7
|
|
| PubChem CID |
158758
|
|
| 外观&性状 |
Off-white to light yellow solid powder
|
|
| 密度 |
1.2±0.1 g/cm3
|
|
| 沸点 |
472.7±45.0 °C at 760 mmHg
|
|
| 熔点 |
133-136ºC
|
|
| 闪点 |
239.7±28.7 °C
|
|
| 蒸汽压 |
0.0±1.2 mmHg at 25°C
|
|
| 折射率 |
1.552
|
|
| LogP |
1.43
|
|
| tPSA |
47.56
|
|
| 氢键供体(HBD)数目 |
1
|
|
| 氢键受体(HBA)数目 |
3
|
|
| 可旋转键数目(RBC) |
4
|
|
| 重原子数目 |
20
|
|
| 分子复杂度/Complexity |
341
|
|
| 定义原子立体中心数目 |
1
|
|
| SMILES |
COC1=C(C=C(C=C1)[C@@H]2CC(=O)NC2)OC3CCCC3
|
|
| InChi Key |
HJORMJIFDVBMOB-GFCCVEGCSA-N
|
|
| InChi Code |
InChI=1S/C16H21NO3/c1-19-14-7-6-11(12-9-16(18)17-10-12)8-15(14)20-13-4-2-3-5-13/h6-8,12-13H,2-5,9-10H2,1H3,(H,17,18)/t12-/m1/s1
|
|
| 化学名 |
(4S)-4-(3-cyclopentyloxy-4-methoxyphenyl)pyrrolidin-2-one
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (9.08 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (9.08 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (9.08 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 30% PEG400+0.5% Tween80+5% propylene glycol:10 mg/L 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.6319 mL | 18.1594 mL | 36.3187 mL | |
| 5 mM | 0.7264 mL | 3.6319 mL | 7.2637 mL | |
| 10 mM | 0.3632 mL | 1.8159 mL | 3.6319 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT05522673 | Terminated Has Results | Drug: 11(R)-rolipram | Depression | National Institute of Mental Health (NIMH) |
February 8, 2023 | Phase 1 |
| NCT00011375 | Completed | Drug: Rolipram | Multiple Sclerosis | National Institute of Neurological Disorders and Stroke (NINDS) |
February 2001 | Phase 2 |
| NCT01215552 | Terminated | Drug: HT-0712 | Healthy Elderly Volunteers | Dart NeuroScience, LLC | September 2010 | Phase 1 |
| NCT00250172 | Completed | Drug: [C-11](R)-rolipram | Dosimetry Healthy |
National Institute of Mental Health (NIMH) |
October 31, 2005 | Phase 1 |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA
 463611831
463611831
