| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 100mg |
|
||
| 1g |
|
||
| 5g |
|
||
| 50g |
|
||
| 100g |
|
||
| Other Sizes |
|
| 靶点 |
- Bacterial 30S Ribosomal Subunit: Inhibits bacterial protein synthesis by binding to the 30S subunit, with a minimum inhibitory concentration (MIC) of 0.06–2 μg/mL against Helicobacter pylori [8]
- Mitochondrial Respiratory Chain Enzymes: Inhibits complex I and III of the mitochondrial electron transport chain (no IC₅₀ reported) [2] - Matrix Metalloproteinases (MMPs): Downregulates MMP-2 and MMP-9 expression in breast cancer cells (no Ki value reported) [5] - Tetracycline-Responsive Transcriptional Regulator (TetR): Binds to TetR to modulate gene expression in inducible systems (no binding affinity value reported) [6,7] |
|---|---|
| 体外研究 (In Vitro) |
1. KRAS突变肺癌细胞的抗增殖活性
- 细胞系:A549(KRAS G12S突变)和H460(KRAS Q61H突变)肺癌细胞。
- 处理方式:多西环素(Doxycycline)(1–10 μg/mL)单独处理或与司美替尼(MEK抑制剂,1 μM)联合处理72小时。
- 结果:
- 单独处理:10 μg/mL 多西环素 使细胞活力降低35–40%(MTT法) [1]
- 联合处理:协同抑制增殖,联合指数(CI)为0.6–0.8;凋亡率较单药组升高2.5倍(TUNEL法) [1] 2. 胶质瘤细胞缺氧诱导细胞死亡的保护作用 - 细胞系:U87和U251人胶质瘤细胞。 - 处理方式:多西环素(5–20 μg/mL)预处理24小时,随后在缺氧环境(1% O₂)中培养48小时。 - 结果: - 缺氧诱导的细胞死亡减少40–50%(Annexin V/PI染色) [2] - 线粒体膜电位(ΔΨₘ)升高30%(JC-1染色),ATP水平升高25%(荧光素酶法) [2] - 缺氧诱导因子1α(HIF-1α)蛋白表达下调60%(Western blot) [2] 3. 乳腺癌干细胞(CSC)表型及上皮-间质转化(EMT)的抑制 - 细胞系:MDA-MB-231和MCF-7乳腺癌细胞。 - 处理方式:多西环素(2–8 μg/mL)处理5–7天。 - 结果: - CSC球形成能力降低50–70%(球形成实验) [5] - CSC标志物(CD44⁺/CD24⁻比例降低45%)和EMT标志物(波形蛋白降低55%,E-钙黏蛋白升高40%)下调(流式细胞术和Western blot) [5] - 细胞迁移能力降低60%(划痕实验) [5] 4. 对幽门螺杆菌的抗菌活性 - 实验方法:肉汤微量稀释法检测临床分离的幽门螺杆菌菌株(n=120)。 - 结果:多西环素 的MIC值范围为0.06–2 μg/mL,92%的菌株对其敏感(MIC ≤ 1 μg/mL) [8] - 药物协同作用:与阿莫西林和克拉霉素联合使用时,幽门螺杆菌根除率较双药治疗提高20%(体外棋盘法) [8] 胶质瘤细胞生长仅受高浓度盐酸多西环素(0.01–10 µg/mL,4 d)的影响[2]。当以 1 µg/mL 及以上的浓度添加到 SVG 细胞中时,盐酸多西环素(0.01–10 µg/mL,24 小时)会减少 MT-CO1 蛋白的量 [2]。盐酸多西环素(100 ng/mL,1 µg/mL;24 小时)可抑制人细胞系增殖[4]。盐酸多西环素(0-250 μM,72 小时)可抑制乳腺癌细胞的细胞活力[5]。 |
| 体内研究 (In Vivo) |
1. KRAS突变肺癌小鼠模型的抗肿瘤疗效
- 动物模型:裸鼠(6周龄雌性)接种A549(KRAS G12S)细胞构建异种移植模型(肿瘤体积约100 mm³)。
- 处理方式:
- 组1:溶媒(0.5%羧甲基纤维素,口服,每日1次) [1]
- 组2:多西环素(50 mg/kg,口服,每日1次) [1] - 组3:司美替尼(25 mg/kg,口服,每日2次) [1] - 组4:多西环素 + 司美替尼(剂量同上) [1] - 处理时长:21天。 - 结果: - 组2:肿瘤生长抑制率(TGI)为28% [1] - 组3:TGI为45% [1] - 组4:TGI为72%;肿瘤重量较溶媒组降低65% [1] 2. 血管型埃勒斯-当洛斯综合征(EDS)小鼠主动脉病变的改善 - 动物模型:Col3a1⁺/⁻小鼠(血管型EDS,8周龄雄性)。 - 处理方式:多西环素(10 mg/kg,口服,每日1次)处理12周。 - 结果: - 主动脉扩张程度降低30%(超声心动图) [3] - 主动脉壁弹性改善(杨氏模量升高25%)(拉伸试验) [3] - 主动脉MMP-9活性降低40%(酶谱法) [3] 3. 大鼠黑质纹状体GDNF表达的调控 - 动物模型:雄性Wistar大鼠(250–300 g),黑质区注射rAAV-Tet-On-GDNF载体。 - 处理方式:多西环素(0.1–1 mg/mL,溶于饮用水)处理4周。 - 结果: - 1 mg/mL 多西环素 使黑质区GDNF mRNA表达升高8倍(qPCR) [6] - GDNF蛋白水平呈剂量依赖性升高;1 mg/mL组较溶媒组升高5倍(Western blot) [6] 在未接受治疗的 HT 小鼠中,盐酸多西环素(口服灌胃;200 或 800 mg/kg;每天一次;3 个月)以剂量依赖性方式降低 MMP-9 活性 [3]。 |
| 酶活实验 |
1. 线粒体呼吸链酶活性检测
- 试剂:U87胶质瘤细胞分离的线粒体组分、NADH(复合物I底物)、琥珀酸(复合物II底物)、细胞色素c(复合物IV底物)。
- 流程:
1. 差速离心法(4°C下800×g离心10分钟,随后10,000×g离心20分钟)分离经多西环素(10 μg/mL)处理24小时的细胞线粒体 [2]
2. 线粒体重悬于检测缓冲液(25 mM Tris-HCl,pH 7.4,5 mM MgCl₂);通过监测340 nm处NADH氧化速率5分钟,检测复合物I活性 [2] 3. 通过监测550 nm处细胞色素c还原速率3分钟,检测复合物III活性 [2] - 结果:多西环素 使复合物I活性降低35%,复合物III活性降低28%(与溶媒组相比) [2] 2. MMP-9酶谱法检测 - 试剂:Col3a1⁺/⁻小鼠主动脉平滑肌细胞(ASMCs)的条件培养基、含0.1%明胶的10% SDS-PAGE凝胶。 - 流程: 1. 多西环素(5 μg/mL)处理ASMCs 48小时,收集条件培养基 [3] 2. 上样培养基(20 μg蛋白)至明胶-SDS-PAGE凝胶,100 V电泳90分钟 [3] 3. 凝胶在复性缓冲液(2.5% Triton X-100)中孵育1小时,随后在发育缓冲液(50 mM Tris-HCl,pH 7.5,5 mM CaCl₂)中37°C孵育过夜 [3] 4. 考马斯亮蓝R-250染色凝胶;通过密度法量化透明带(MMP-9活性) [3] - 结果:多西环素 使MMP-9活性降低40%(与溶媒组相比) [3] |
| 细胞实验 |
细胞活力测定[2]
细胞类型: LNT-229、G55 和 U343 胶质瘤细胞 测试浓度: 0.01、0.1、1 或 10 µg/ mL 孵育时间:4 天 实验结果:仅在高浓度(10 µg)神经胶质瘤细胞生长/ml 时受影响。 细胞活力测定[2] 细胞类型: SVG 细胞 测试浓度: 0.01、0.1、1 或 10 µg/mL 孵育持续时间:24 小时 实验结果:MT-CO1 蛋白含量在浓度为 1 µg/mL 及更高时减少。 细胞增殖测定 [4] 细胞类型: MCF 12A、293T 细胞 测试浓度: 100 ng/mL,1 µg /mL 孵育持续时间:96 小时 实验结果:1 µg/mL 导致 MCF 12A 和 293T 细胞增殖减弱。 细胞活力测定[5] 细胞类型: MCF-7、MDA-MB-468 细胞 测试浓度: 0- 250 μM 孵育时间:72小时 实验结果:对乳腺癌细胞MCF-7和MCF-7的抑制作用MDA-MB-468的IC50值分别为11.39μM和7.13μM,呈剂量依赖性。 1. 缺氧诱导胶质瘤细胞死亡检测 - 流程: 1. 96孔板接种U87胶质瘤细胞(5×10³细胞/孔);37°C、5% CO₂培养24小时 [2] 2. 更换为含多西环素(0、5、10、20 μg/mL)的新鲜培养基,继续培养24小时 [2] 3. 平板转移至缺氧培养箱(1% O₂、5% CO₂、94% N₂)培养48小时 [2] 4. MTT法检测细胞活力(加入20 μL MTT溶液,孵育4小时;DMSO溶解甲瓒,570 nm处测吸光度) [2] 5. Annexin V-FITC/PI染色检测凋亡(细胞与Annexin V和PI孵育15分钟;流式细胞术分析) [2] - 结果:20 μg/mL 多西环素 使缺氧环境下细胞活力升高50%,凋亡率降低45% [2] 2. 乳腺癌干细胞球形成实验 - 流程: 1. MDA-MB-231细胞在含EGF(20 ng/mL)和bFGF(10 ng/mL)的无血清培养基(SFM)中培养7天,形成原代球 [5] 2. 球解离为单细胞;接种于6孔板(1×10³细胞/孔),培养基为含多西环素(0、2、4、8 μg/mL)的SFM [5] 3. 孵育10天;计数直径>50 μm的球 [5] 4. 流式细胞术分析CSC标志物(CD44/CD24)(细胞用抗CD44-PE和抗CD24-FITC抗体染色;流式细胞仪检测) [5] - 结果:8 μg/mL 多西环素 使球数量减少70%,CD44⁺/CD24⁻细胞比例降低45% [5] |
| 动物实验 |
1. KRAS突变肺癌异种移植小鼠模型 - 方案: 1. 制备A549细胞(1×10⁷个细胞/mL PBS); 1. 将 0.1 mL 溶液皮下注射到裸鼠(6 周龄雌性)右侧腹部 [1]
2. 当肿瘤体积达到约 100 mm³ 时,将小鼠随机分为 4 组(每组 n=6)[1] 3. 每日给药,持续 21 天: - 赋形剂:0.5% 羧甲基纤维素(100 μL,灌胃)[1] - 多西环素:50 mg/kg,溶于赋形剂(100 μL,灌胃)[1] - 塞鲁米替尼:25 mg/kg,溶于 DMSO(100 μL,灌胃,每日两次)[1] - 联合用药:多西环素(50 mg/kg)+ 塞鲁米替尼(25 mg/kg,每日两次)[1] 4. 每隔 3 天测量一次肿瘤体积(V = 长 × 宽² / 2)。天;安乐死后称量肿瘤[1] 5. 收集肿瘤组织进行蛋白质印迹分析(分析 Ki67、cleaved caspase-3)[1] 2. 血管型 EDS 小鼠模型 - 实验方案:1. 使用 8 周龄雄性 Col3a1⁺/⁻ 小鼠(每组 n=8);分为载体组和强力霉素组[3] 2. 强力霉素组:将 10 mg/kg 强力霉素溶于饮用水中(自由饮用),持续 12 周[3] 3. 载体组:普通饮用水[3] 4. 在基线和第 12 周进行腹主动脉超声检查,测量主动脉直径[3] 5. 安乐死小鼠;分离主动脉进行拉伸试验(测量杨氏模量)和酶谱分析(检测 MMP-9 活性)[3] 动物/疾病模型: 6 月龄雌性杂合子 Col3a1 缺陷 (HT) 小鼠 [3] 剂量: 200 或 800 mg/kg 给药途径: 口服(灌胃);200 或 800 mg/kg;每日一次;持续 3 个月 实验结果: MMP-9 活性呈剂量依赖性降低。 |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
口服多西环素后几乎完全吸收,生物利用度为73-95%。口服500 mg后,4小时达到血药浓度峰值(Cmax),为15.3 mg/L。正常成年志愿者口服200 mg后,2小时血药浓度峰值平均为2.6 mcg/mL,24小时降至1.45 mcg/mL。虽然高脂饮食会降低血药浓度峰值和吸收率,但这种影响在临床上并不显著。 包括多西环素在内的四环素类药物在肝脏中被胆汁浓缩,并以高浓度和生物活性形式经尿液和粪便排出。肌酐清除率约为75 mL/min的个体,多西环素经肾脏排泄的比例约为40%/72小时。对于肌酐清除率低于 10 mL/min 的个体,该百分比可能低至 1-5%/72 小时。 目前相关信息有限。 对 44 名 2 至 18 岁儿科患者接受标准静脉和口服给药后,多西环素的稀疏浓度-时间数据进行群体药代动力学分析,结果显示异速缩放清除率 (CL) 为 3.27 至 3.58 L/h/70 kg。 已证实多西环素药代动力学对肾功能不全相对不敏感,这似乎与药物扩散到小肠腔内导致粪便排泄增加有关。活性抗生素的肾清除率为……多西环素为20毫升/分钟……。 无论静脉注射还是口服,多西环素的血清浓度均相同。每日多次静脉注射200毫克后,血清浓度在5-6微克/毫升和1-2微克/毫升之间波动,高于大多数易感病原体的最低抑制浓度。 尿液pH值较高时,多西环素的尿排泄量增加。与酸性治疗相比,碱性治疗使受试者尿中四环素的累积排泄量增加了 24%(P< 0.05),多西环素的累积排泄量增加了 54%(P < 0.05)。肾清除率……碱性治疗期间增加……。 ……口服后吸收比其他四环素类药物更完全……在血浆中,其蛋白结合率约为90%,是所有四环素类药物中最高的。 有关多西环素(共20项)的更多吸收、分布和排泄(完整)数据,请访问HSDB记录页面。 代谢/代谢物 相关信息有限。 多西环素主要以无活性结合物或螯合物的形式经粪便排泄(高达90%)。 虽然之前有研究表明多西环素部分在肝脏代谢,但最近的研究表明该药物并不在肝脏代谢。但在肠道中会通过螯合作用部分失活。 生物半衰期 目前信息有限。 多西环素:排泄途径:肝脏、肾脏;正常半衰期:20 小时;维持剂量间隔:12-24 小时。 多西环素为长效药物,首次给药后血清半衰期为 15-17 小时,治疗第 4 天后约为 22 小时。 肾功能正常的患者单次服用多西环素后血清半衰期为 14-17 小时,多次给药后为 22-24 小时。在严重肾功能损害的患者中,单次服用多西环素后,其血清半衰期据报道为 18-26 小时,多次服用后为 20-30 小时。血液透析患者的多西环素血清半衰期似乎没有改变。肾功能正常的患者,单次口服或静脉注射多西环素后,约 20-26% 的药物在 48 小时内以活性药物的形式经尿液排出,20-40% 的药物经粪便排出。肌酐清除率低于 10 ml/分钟的患者,72 小时内经尿液排出的多西环素比例可能降至约 1-5%。吸收:多西环素的口服生物利用度约为 90%(在人体中)。口服100 mg后,2-3小时达到血浆峰浓度(Cₘₐₓ) 2-4 μg/mL [4,8] - 食物摄入会略微降低吸收(约10%),但无需调整剂量 [8] - 分布: - 分布容积(Vd)为0.7-1.0 L/kg;广泛分布于组织(肺、肝、肾、肿瘤)[4] - 血浆蛋白结合率约为80-90% [4] - 代谢: - 肝脏代谢极少;大部分药物以原形排出 [4] - 排泄: - 主要经粪便(40-50%)和尿液(30-40%)排泄;末端半衰期(t₁/₂)为12-22小时 [4,8] |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
多西环素与罕见的肝损伤病例相关,通常在开始治疗后 1 至 2 周内出现,有时既往服用该药未出现肝损伤。肝损伤类型从肝细胞性到胆汁淤积性不等,最常见的可能是混合型。起病通常突然,并可能伴有超敏反应症状,例如发热、皮疹和嗜酸性粒细胞增多(药物反应伴嗜酸性粒细胞增多和全身症状综合征,DRESS 综合征)。恢复通常迅速,通常在 4 至 6 周内完全恢复。然而,已有口服多西环素导致严重且持续性胆汁淤积性肝损伤的报道。尽管化学结构相似,适应症和用途也相似,但与米诺环素相关的自身免疫样肝炎尚未与多西环素联系起来,这可能是因为多西环素的使用频率较低,且通常采用低剂量、长期治疗方案。大剂量静脉注射多西环素可引起急性脂肪肝,其症状与静脉注射四环素引起的脂肪肝相似,尤其是在孕妇等易感人群中。然而,这种损伤非常罕见。尽管如此,鉴于上述原因,应尽量减少肠外多西环素治疗的疗程和剂量。 可能性评分:B(极有可能但罕见地引起临床上明显的肝损伤)。 妊娠期和哺乳期的影响 ◉ 哺乳期用药概述 一些综述指出,由于四环素类药物可能导致婴儿牙釉质染色或沉积于骨骼,因此哺乳期禁用。然而,仔细查阅现有文献表明,哺乳期短期使用多西环素不太可能造成危害,因为乳汁中的药物浓度较低,且婴儿对药物的吸收会受到母乳中钙的抑制。目前认为,8岁以下儿童使用多西环素疗程不超过21天是可以接受的。但作为一项理论上的预防措施,哺乳期妇女应避免疗程过长(超过21天)或重复疗程。监测婴儿皮疹情况以及可能对胃肠道菌群产生的影响,例如腹泻或念珠菌病(鹅口疮、尿布疹)。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对哺乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白质结合 虽然现有信息有限,但四环素类药物与血浆蛋白的结合程度各不相同。 相互作用 口服硫酸亚铁(200-600毫克)会干扰胃肠道对多西环素的吸收,反之亦然,导致抗生素和铁盐的血清浓度分别降低。如果必须同时给药,患者应在服用铁剂后 3 小时或服用铁剂前 2 小时服用多西环素。 据报道,多西环素与氢氧化铝存在相互作用。 同时服用卡马西平(得理通)、苯妥英钠(大仑丁)或巴比妥类药物会加速多西环素的肝脏代谢,从而缩短其半衰期。 同时使用四环素类药物和皮质类固醇可能导致二重感染。……服用四环素类药物和利尿剂的患者血尿素氮水平升高。四环素类药物不应与其他可能具有肝毒性的药物同时服用。 /四环素类/ 有关多西环素(共11项)的更多相互作用(完整)数据,请访问HSDB记录页面。 非人类毒性值 小鼠口服LD50:1007.45 mg/kg 小鼠静脉注射LD50:204-222.5 mg/kg - 体外毒性:- 多西环素(浓度高达20 μg/mL)不会对正常人成纤维细胞产生显著的细胞毒性(MTT法检测细胞活力>90%)[4] - 高浓度(>50 μg/mL)可抑制正常肺上皮细胞(BEAS-2B)增殖达30%[1] - 体内毒性:- 小鼠接受多西环素治疗(50 mg/kg/天,持续21天)后,未见显著变化观察了体重、肝功能(ALT、AST)或肾功能(BUN、肌酐)的变化[1] - 在用多西环素(饮用水中浓度为1 mg/mL,持续4周)治疗的大鼠中,观察到轻度胃肠道刺激(食物摄入量减少10%),但停药后症状消退[6] - 人体副作用: - 常见副作用包括恶心(15%)、腹泻(10%)和光敏性(5%)[8] - 罕见副作用:肝功能障碍(发生率<0.1%)和超敏反应[8] |
| 参考文献 |
|
| 其他信息 |
治疗用途
抗生素,四环素 对革兰氏阳性菌的抗菌效力约为四环素的两倍,但对草绿色链球菌的抗菌效力可达四环素的十倍。此外,对其他四环素类药物耐药的粪链球菌菌株可能对多西环素敏感。 成人多西环素的剂量为:最初24小时内每12小时服用100毫克,之后每日一次,或在严重感染时每日两次。 8岁以上儿童应每日服用4-5毫克/公斤体重,分两次服用,间隔12小时,第一天服用两次;之后,每日服用一半剂量。 由于多西环素可与食物或牛奶同服,不会显著降低其活性或影响吸收,因此其对金属离子的亲和力可能不如其他四环素类药物。 有关多西环素(共27种)的更多治疗用途(完整)数据,请访问HSDB记录页面。 药物警告 孕妇或哺乳期妇女禁用四环素类药物。除非有充分的理由,否则妇女和 8 岁以下儿童禁用。 孕妇用药可能导致其后代牙齿变色。8 岁以下儿童可能易感。四环素类药物在妊娠期间沉积于骨骼中。早产儿用此类药物治疗后,骨骼生长抑制率达 40%。四环素类药物对孕妇构成特殊危险,可能导致肝损伤,尤其是在用于治疗肾盂肾炎时,这种情况在孕妇中较为常见,甚至已有死亡病例发生。四环素类药物之间交叉致敏现象很常见。有关多西环素(共12条)的更多药物警告(完整)数据,请访问HSDB记录页面。药效学:多西环素和其他四环素类药物主要为抑菌剂,其抗菌作用被认为是通过抑制蛋白质合成实现的。它们抑制细菌生长或使其处于静止期。四环素类药物对多种革兰氏阳性菌和革兰氏阴性菌具有抗菌谱。这些微生物对四环素类药物的交叉耐药性很常见。由于多西环素是一种高度亲脂性药物,因此可以穿过靶分子的多层膜。多西环素具有良好的细胞内渗透性,对多种细菌具有抑菌活性。多西环素还具有抗寄生虫和抗炎作用。其抗炎作用已在多种炎症性皮肤病(如大疱性皮炎和酒渣鼻)中得到研究。 - 作用机制: - 抗菌:与细菌 30S 核糖体亚基结合,阻断氨酰 tRNA 与 A 位点的结合,从而抑制蛋白质合成 [8] - 抗肿瘤:抑制线粒体功能,减少癌症干细胞的自我更新,并下调 MMPs 以抑制肿瘤生长和转移 [1,5] - 基因调控:作为 TetR 的配体,诱导或抑制 Tet 诱导系统中的基因表达(例如,调节大脑中的 GDNF 表达)[6,7] - 临床疗效: - 幽门螺杆菌根除:与阿莫西林和质子泵抑制剂 (PPI) 联合使用,以多西环素为基础的三联疗法可达到 80-85% 的根除率(优于克拉霉素耐药菌株的克拉霉素疗法) [8] - 癌症治疗:多西环素(50 mg/kg/天)可增强 MEK 抑制剂在 KRAS 突变型肺癌中的疗效,且不增加毒性[1] - 研究应用: - 用于四环素诱导型转基因小鼠模型,以实现组织特异性和时间依赖性基因表达控制[7] |
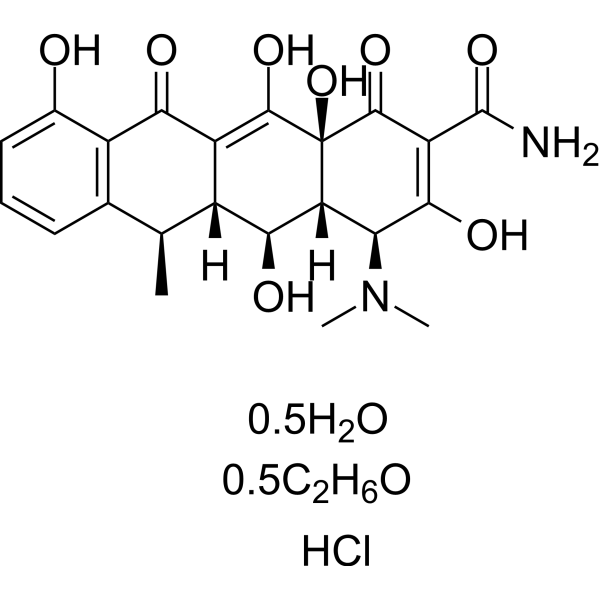
| 分子式 |
C24H32CL2N2O10
|
|---|---|
| 分子量 |
579.42
|
| 精确质量 |
444.153
|
| 元素分析 |
C, 53.86; H, 5.70; Cl, 6.91; N, 5.46; O, 28.07
|
| CAS号 |
24390-14-5
|
| 相关CAS号 |
Doxycycline;564-25-0;Doxycycline hydrochloride;10592-13-9;Doxycycline monohydrate;17086-28-1;Doxycycline calcium;94088-85-4
|
| PubChem CID |
54671203
|
| 外观&性状 |
White to yellow solid powder
|
| 沸点 |
685.2ºC at 760 mmHg
|
| 熔点 |
206-209?C (dec.)
|
| 闪点 |
368.2ºC
|
| 蒸汽压 |
1.03E-19mmHg at 25°C
|
| LogP |
2.243
|
| tPSA |
392.7
|
| 氢键供体(HBD)数目 |
6
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
32
|
| 分子复杂度/Complexity |
956
|
| 定义原子立体中心数目 |
6
|
| SMILES |
C[C@@H]1[C@H]2[C@@H]([C@H]3[C@@H](C(=O)C(=C([C@]3(C(=O)C2=C(C4=C1C=CC=C4O)O)O)O)C(=O)N)N(C)C)O
|
| InChi Key |
SGKRLCUYIXIAHR-AKNGSSGZSA-N
|
| InChi Code |
InChI=1S/C22H24N2O8/c1-7-8-5-4-6-9(25)11(8)16(26)12-10(7)17(27)14-15(24(2)3)18(28)13(21(23)31)20(30)22(14,32)19(12)29/h4-7,10,14-15,17,25-27,30,32H,1-3H3,(H2,23,31)/t7-,10+,14+,15-,17-,22-/m0/s1
|
| 化学名 |
(4S,4aR,5S,5aR,6R,12aR)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4H-tetracene-2-carboxamide
|
| 别名 |
Doxy-Lemmon; Vivox; DTXSID80992212; 4-(Dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-3,4,4a,5,5a,6,12,12a-octahydrotetracene-2-carboximidic acid; 7164-70-7; RefChem:1070088; ...; 24390-14-5; Doxycycline Hyclate
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮和光照。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~240 mg/mL (~467.89 mM)
H2O : ~125 mg/mL (~243.69 mM) |
|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 3 mg/mL (5.85 mM) (饱和度未知) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
*生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 3 mg/mL (5.85 mM) (饱和度未知) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: 20 mg/mL (38.99 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7259 mL | 8.6293 mL | 17.2586 mL | |
| 5 mM | 0.3452 mL | 1.7259 mL | 3.4517 mL | |
| 10 mM | 0.1726 mL | 0.8629 mL | 1.7259 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Enhanced Dermatological Care to Reduce Rash and Paronychia in Epidermal Growth Factor Receptor (EGRF)-Mutated Non-Small Cell Lung Cancer (NSCLC) Treated First-line With Amivantamab Plus Lazertinib
CTID: NCT06120140
Phase: Phase 2 Status: Recruiting
Date: 2024-10-26
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
