| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|
| 靶点 |
PKA; PDE
In vitro activity: Bucladesine sodium (also known as Dibutyryl-cAMP) is a cell-permeable PKA activator and a cAMP analog that mimics the action of endogenous cAMP. It is a cyclic nucleotide derivative (structurally similar to cAMP) and is also a phosphodiesterase inhibitor. Dibutyryl-cAMP preferentially activates cAMP-dependent protein kinase. The products releaes butyrate due to intracellular and extracellular esterase action. Butyrate was shown to have distinct biological effects. The compound is used in a wide variety of research applications because it mimics cAMP and can induce normal physiological responses when added to cells in experimental conditions. Kinase Assay: Bucladesine sodium (also known as Dibutyryl-cAMP) is a cell-permeable PKA activator and a cAMP analog that mimics the action of endogenous cAMP. It is a cyclic nucleotide derivative (structurally similar to cAMP) and is also a phosphodiesterase inhibitor. Dibutyryl-cAMP preferentially activates cAMP-dependent protein kinase. |
|---|---|
| 体外研究 (In Vitro) |
体外活性:布克拉地辛钠(也称为二丁酰-cAMP)是一种细胞渗透性 PKA 激活剂和模拟内源性 cAMP 作用的 cAMP 类似物。它是一种环核苷酸衍生物(结构与cAMP相似),也是一种磷酸二酯酶抑制剂。二丁酰-cAMP 优先激活 cAMP 依赖性蛋白激酶。该产品由于细胞内和细胞外酯酶的作用而释放丁酸盐。丁酸盐被证明具有独特的生物效应。该化合物可用于多种研究应用,因为它模仿 cAMP,并且在实验条件下添加到细胞中时可以诱导正常的生理反应。激酶测定:布克拉地辛钠(也称为二丁酰-cAMP)是一种细胞渗透性 PKA 激活剂和模拟内源性 cAMP 作用的 cAMP 类似物。它是一种环核苷酸衍生物(结构与cAMP相似),也是一种磷酸二酯酶抑制剂。二丁酰-cAMP 优先激活 cAMP 依赖性蛋白激酶。
研究开发了一种用于局部给药的含 Bucladesine (DB-cAMP) 的无水乳剂。测定了 Bucladesine (DB-cAMP) 在不同溶剂中的溶解度。其在疏水性溶剂中溶解度差,但在丙二醇、乙二醇和N-甲基吡咯烷酮中溶解度良好。该化合物溶解在N-甲基吡咯烷酮中时保持稳定,14天内未观察到水解。[3] |
| 体内研究 (In Vivo) |
雄性大鼠双侧输注 10 mM 或 100 mM 布克拉地辛,结果显示与对照组相比,逃避潜伏期和行走距离显着缩短(表明空间记忆得到改善)。雄性大鼠在接受 0.5 mg 尼古丁后几分钟内注射 1 或 5 mM 布氯地辛,保留空间记忆的能力会增强 [1]。当布克拉地辛(10 mM/侧)和尼古丁(0.5 mM/侧)联合使用时,CA1 区域的 ChAT 和 VAChT 免疫反应性显着升高。此外,使用尼古丁和低剂量布克拉地辛治疗时,ChAT 和 VAChT 免疫染色的光密度和数量显着增加。大鼠的逃避延迟和行走距离减少[2]。将水溶液施用于皮肤切除部位后,布克拉地辛几乎很快被完全吸收。与全层磨损大鼠模型相比,在没有皮肤脱皮的情况下,布克拉地辛的吸收更慢但更快[3]。在花生四烯酸引起的小鼠耳水肿模型中,布克拉地辛(含1.5%乳剂,单次或多次注射)可显着减轻炎性水肿[4]。
将3%的 布卡地辛 水溶液 (30 mg/只) 局部涂抹于皮肤完整的大鼠后,血浆浓度极低,系统生物利用度仅为 1.2 ± 0.5%。 [2] 将相同的3%水溶液涂抹于全层皮肤擦伤部位时,布卡地辛 吸收非常迅速 (达峰时间 0.5 小时),且几乎被完全吸收(生物利用度:93.1 ± 34.3%)。 [2] 将3%水溶液涂抹于剥离了角质层的皮肤后,吸收也很快(达峰时间 0.5 小时),生物利用度为 63.5 ± 14.4%,但峰值血药浓度约为全层擦伤模型的四分之一。 [2] 3%的 布卡地辛 聚乙二醇软膏能提供持续释放。涂抹于全层擦伤部位时,生物利用度为 31.8 ± 27.4%,与水溶液相比达峰时间延迟。涂抹于剥离角质层的皮肤时,PEG软膏的生物利用度为 10.6 ± 4.1%。 [2] 3%的 布卡地辛 凡士林软膏在剥离角质层(生物利用度:0.6 ± 1.4%)和全层擦伤模型(生物利用度:3.7 ± 0.7%)中的吸收均非常低。 [2] 将 布卡地辛 原料药粉末 (30 mg) 局部涂抹于剥离角质层的皮肤后,吸收迅速且几乎完全(生物利用度:84.3 ± 18.4%),峰值血药浓度高于水溶液。 [2] |
| 酶活实验 |
PKA试验[4]
用10 mM磷酸钠缓冲液,pH 7.4, 0.15 M NaC1洗涤细胞2次,然后用1 ml相同的缓冲液从培养板上刮下。离心收集细胞,在细胞匀浆缓冲液(50 mMTris-HC1, pH 7.4, 1 mM EDTA, 1 mM二硫苏糖醇(DTT), 50 mM胰肽,0.1 mM苯基甲基磺酰氟)中进行短暂超声匀浆。在4°c, 14000 rpm的微离心机中离心20 mm,去除颗粒部分。采用froskoski(1983)的方法,使用合成肽底物Leu-Arg-ArgAla-Ser-Leu-Gly (Kemptide),在上清液中测量PKA活性。反应混合物为50 ~。含有细胞裂解液,终浓度为25 mM Tris-HC1缓冲液(pH7.4), 5 mM醋酸镁,5 mM DTT, 5 mM cAMP, 20,~iMKemptide, 0.25 mM异丁基甲基黄嘌呤,0.1 mM [y- 32P I ATP (200 cpm/pmol),当添加20,uM PKA肽抑制剂5-24时。在30°温度下,用50jtl的7.5 mm磷酸终止反应10 mm。将50微升反应混合物放在P81过滤器上,用75 mM磷酸洗涤5次,并按前面描述的计数。PKA肽抑制剂5-24存在与不存在时的活性差异用于计算PKA活性。 PKC检测[4] 按照pka实验的描述制备细胞裂解物。反应液为50 j.el,终浓度为25 mM Tris-HC1缓冲液(pH 7.4), 5 mM醋酸镁,5 mM DTT, 20 ~。tM合成底物(Pro-Leu-Ser-Arg-Thr-Leu-Ser-Val-Ala-Ala-LysLys), 0.25 mM异丁基甲基黄嘌呤,0.1 mM [y32p] ATP (200 cpm/pmol)。反应在30°C下孵育10 mm,用磷酸终止,并按照PKA试验的描述进行分析。作为对照,特异性PKC肽抑制剂19-36,在20。用tM对细胞提取物的活性有90%以上的抑制作用。 |
| 细胞实验 |
囊泡乙酰胆碱转运体(VAChT)基因和胆碱乙酰转移酶(ChAT)基因构成胆碱能基因座。我们研究了环腺苷酸依赖性蛋白激酶(PKA)在大鼠嗜铬细胞瘤细胞系PC12和PC12 PKA缺陷突变体中对这些基因的协同调节。二丁基环腺苷酸(dbcAMP)处理PC12细胞后,ChAT和VAChT mRNA均增加了约四倍。dbcAMP也能提高ChAT和PKA的活性。PKA缺陷细胞系中ChAT和VAChT mRNA的基础水平均比野生型PC12细胞低约6倍,并且通过添加dbcAMP诱导不到两倍。PKA的特异性抑制剂H-89和H-9将ChAT和VAChT mRNA水平降低到未处理细胞的约三分之一,ChAT活性降低到未治疗PC12细胞的约四分之一。激活PKA II型而不是PKA I型,使ChAT活性增加约三倍。报告基因构建体的分析表明PKA影响胆碱能基因位点上游位点的基因转录。这些结果表明,ChAT和VAChT基因的表达在转录水平上受到协同调节,特异性涉及PKA II的信号通路在这一过程中发挥着重要作用[5]。
|
| 动物实验 |
局部应用布克拉地辛(5%)溶液时,在花生四烯酸刺激前60分钟,分别将20 μl药物或赋形剂溶液涂抹于左右耳外表面。通过在左耳外表面涂抹20 μl花生四烯酸(Sigma-Aldrich,慕尼黑,德国;5%丙酮溶液)诱导炎症反应。右耳仅用丙酮处理,以确定耳厚度的个体差异。未接受过任何处理的雄性瑞士白化小鼠
大鼠经皮吸收研究:使用8周龄雄性Sprague-Dawley大鼠。实验前16小时剃除腹部毛发。大鼠麻醉并固定。制备三种皮肤状态:1)正常完整皮肤。2)剥离皮肤:通过反复胶带剥离(20次)去除角质层。 3) 全层皮肤擦伤:用剪刀切除腹部皮肤。将一个塑料容器(内径 3 cm)固定在施药部位。将布地辛(每只大鼠 30 mg)以 3% 水溶液、3% PEG 软膏、3% 凡士林软膏、散装粉末或各种粉末制剂(PW-1、PW-2、PW-3)的形式涂抹于皮肤上。密封容器。分别于施药后 0.5、1、2、4 和 8 小时从颈静脉采集血样。分离血浆并冷冻保存直至分析。[2] 静脉给药用于药代动力学参数测定:大鼠单次静脉注射 3 mg 布地辛水溶液。定期采集血样,持续 8 小时,以确定基线药代动力学参数。[2] |
| 药代性质 (ADME/PK) |
大鼠单次静脉注射布地辛(3 mg)后,血浆浓度呈单指数下降。[2]消除速率常数(ke)为 8.24 ± 1.25 h⁻¹。[2]生物半衰期为 5.14 ± 0.81 分钟。[2]局部用药后的绝对生物利用度计算为经皮给药途径的血浆浓度-时间曲线下面积(AUC)与静脉给药途径的 AUC 之比。[2]将 3% 水溶液应用于正常皮肤、去皮皮肤和全层皮肤擦伤后,其生物利用度分别为 1.2 ± 0.5%、63.5 ± 14.4% 和 93.1 ± 34.3%。 [2]
将3%聚乙二醇(PEG)软膏涂抹于正常皮肤、去皮皮肤和全层皮肤擦伤处,其生物利用度分别为3.2 ± 0.8%、10.6 ± 4.1%和31.8 ± 27.4%。[2] 将3%凡士林软膏涂抹于正常皮肤、去皮皮肤和全层皮肤擦伤处,其生物利用度分别为0.0 ± 0.1%、0.6 ± 1.4%和3.7 ± 0.7%。[2] 将散装粉末和配制粉末(PW-1、PW-2、PW-3)涂抹于去皮皮肤,其生物利用度分别为84.3 ± 18.4%、13.4 ± 8.1%、2.9 ± 1.9%和13.7 ± 7.3%。 [2] |
| 毒性/毒理 (Toxicokinetics/TK) |
文献指出,布克拉地辛(DB-cAMP)作为伤口愈合的局部治疗药物,其安全性极佳,此前已在临床上成功应用。临床应用中未发现PDE4抑制剂类不良反应(例如恶心等胃肠道反应)。然而,其制剂的一个挑战是水解可能产生丁酸,从而导致难闻的气味。本研究开发的无水乳剂旨在防止水解,并在观察期内保持稳定,未出现水解或气味加重的迹象。[3]
|
| 参考文献 |
|
| 其他信息 |
布地辛(N⁶,2′-O-二丁酰环状3′,5′腺苷单磷酸钠,DBcAMP)可有效治疗慢性皮肤溃疡,包括褥疮。[2] 研究表明,布地辛经皮吸收在完整皮肤上几乎可以忽略不计,但在受损皮肤上则显著,这符合其治疗溃疡的预期用途。[2] 制剂载体对经受损皮肤吸收的速率和程度有显著影响。聚乙二醇(PEG)软膏可提供持续释放特性,这有利于最大限度地减少高峰浓度可能引起的全身不良反应。[2] 研究结果支持临床观察,即由于其更优的药物释放特性,PEG软膏在治疗褥疮方面可能比凡士林软膏具有更好的疗效。[2]
|
| 分子式 |
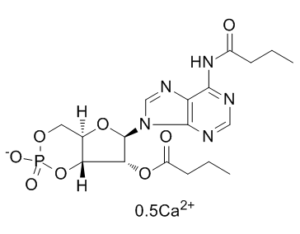
C18H24CAN5O8P
|
|
|---|---|---|
| 分子量 |
509.463624000549
|
|
| 精确质量 |
976.219
|
|
| CAS号 |
938448-87-4
|
|
| 相关CAS号 |
Bucladesine sodium;16980-89-5; 362-74-3
|
|
| PubChem CID |
44514776
|
|
| 外观&性状 |
White to off-white solid
|
|
| tPSA |
328Ų
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
22
|
|
| 可旋转键数目(RBC) |
16
|
|
| 重原子数目 |
65
|
|
| 分子复杂度/Complexity |
751
|
|
| 定义原子立体中心数目 |
8
|
|
| SMILES |
CCCC(=O)NC1=C2C(=NC=N1)N(C=N2)[C@H]3[C@@H]([C@H]4[C@H](O3)COP(=O)(O4)[O-])OC(=O)CCC.CCCC(=O)NC1=C2C(=NC=N1)N(C=N2)[C@H]3[C@@H]([C@H]4[C@H](O3)COP(=O)(O4)[O-])OC(=O)CCC.[Ca+2]
|
|
| InChi Key |
DRYMTGFYEAYJQR-NGVPHMJWSA-N
|
|
| InChi Code |
InChI=1S/2C18H24N5O8P.Ca/c2*1-3-5-11(24)22-16-13-17(20-8-19-16)23(9-21-13)18-15(30-12(25)6-4-2)14-10(29-18)7-28-32(26,27)31-14;/h2*8-10,14-15,18H,3-7H2,1-2H3,(H,26,27)(H,19,20,22,24);/t2*10-,14-,15-,18-;/m11./s1
|
|
| 化学名 |
calcium;[(4aR,6R,7R,7aR)-6-[6-(butanoylamino)purin-9-yl]-2-oxido-2-oxo-4a,6,7,7a-tetrahydro-4H-furo[3,2-d][1,3,2]dioxaphosphinin-7-yl] butanoate
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
配方 1 中的溶解度: ≥ 2.5 mg/mL (5.12 mM) (饱和度未知) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。
例如,若需制备1 mL的工作液,可将100 μL 25.0 mg/mL澄清DMSO储备液加入到400 μL PEG300中,混匀;然后向上述溶液中加入50 μL Tween-80,混匀;加入450 μL生理盐水定容至1 mL。 *生理盐水的制备:将 0.9 g 氯化钠溶解在 100 mL ddH₂O中,得到澄清溶液。 配方 2 中的溶解度: ≥ 2.5 mg/mL (5.12 mM) (饱和度未知) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 例如,若需制备1 mL的工作液,可将 100 μL 25.0 mg/mL澄清DMSO储备液加入900 μL 20% SBE-β-CD生理盐水溶液中,混匀。 *20% SBE-β-CD 生理盐水溶液的制备(4°C,1 周):将 2 g SBE-β-CD 溶解于 10 mL 生理盐水中,得到澄清溶液。 View More
配方 3 中的溶解度: ≥ 2.5 mg/mL (5.12 mM) (饱和度未知) in 10% DMSO + 90% Corn Oil (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液。 配方 4 中的溶解度: 10% DMSO +ddH2O: 30 mg/mL 配方 5 中的溶解度: 110 mg/mL (225.22 mM) in PBS (这些助溶剂从左到右依次添加,逐一添加), 澄清溶液; 超声助溶. 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9629 mL | 9.8143 mL | 19.6286 mL | |
| 5 mM | 0.3926 mL | 1.9629 mL | 3.9257 mL | |
| 10 mM | 0.1963 mL | 0.9814 mL | 1.9629 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Anti-inflammatory effect of 0.5 and 1.5% bucladesine cream given(a)3h before administration of arachidonic acid or given(b)twice, i.e., 7 and 3h before administration of arachidonic acid.Arch Dermatol Res.2012 May;304(4):313-7. |
Anti-inflammatory effect of 5% bucladesine given 1h before administration of arachidonic acid.Arch Dermatol Res.2012 May;304(4):313-7. |
Anti-inflammatory effect of 2.5% ketoprofen gel given 3h before administration of arachidonic acid.Arch Dermatol Res.2012 May;304(4):313-7. |
 Kiss2 peptide acetate
Kiss2 peptide acetate
 Rp-8-Br-cAMPS sodium
Rp-8-Br-cAMPS sodium
 8-Bromo-cAMP
8-Bromo-cAMP
 Sp-8-Br-cAMPS
Sp-8-Br-cAMPS
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA



 463611831
463611831
