| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
Traditional Cytotoxic Agents
|
|---|---|
| 体外研究 (In Vitro) |
Mipsagargin (G-202) 对产生 PSMA 的 LNCaP 细胞 (IC50=5351 nM) 的效力是对不产生 PSMA 的 TSU 细胞 (IC50=191 nM) 的 57 倍[2]。
此外,Mipsagargin (G202)在人血浆中对水解完全稳定;事实上,水解Mipsagargin (G202)的能力是相对PSMA特异性的,因为G202在与其他多种纯化蛋白酶孵育后并没有明显的水解[2]。为了确定G202中的5个氨基酸肽是否可以阻止非特异性细胞摄取,我们将psma -不产生TSU的膀胱癌细胞加载钙敏感染料,并将细胞暴露于12adt - β asp (200 nM)或G202 (5 mM)(6)。将psma -不产生TSU的膀胱癌细胞暴露于12adt - β asp可使细胞内Ca2+快速升高至400 nM(图3E)。相比之下,将相同的细胞暴露于25倍浓度的G202时,细胞内钙没有明显增加(图3F)。 为了证实Mipsagargin (G202)对产生psma的细胞具有选择性毒性,我们比较了Mipsagargin (G202)与不水解psma的12adt β asp - gluγ γ gluu - asp在产生psma的LNCaP人前列腺癌细胞系和不产生psma的TSU细胞系中的细胞毒作用。在本实验中,在细胞增殖实验中,非水解前药抑制50%生长的浓度(IC50)在两种细胞系的微摩尔范围内(图3G)。相比之下,G202对不产生psma的TSU细胞的IC50比产生psma的LNCaP细胞高57倍(图3G)。最后,g202诱导的LNCaP细胞生长抑制可以通过添加2-(磷甲乙酯)-戊二酸(PMPA)来阻断,PMPA是一种有效的PSMA抑制剂,进一步证实了PSMA需要体外前药激活[2] |
| 体内研究 (In Vivo) |
就其本身而言,mipagargin (G-202;56个毫克/公斤;每天两次;49 d)可显著(>50%)抑制肿瘤生长。当每日添加口服HDAC4抑制剂Tasquinimod时,这种回归趋于稳定[1]。
在30天的过程中,完整小鼠用Mipsagargin (G202) (56 mg/kg/天,连续3天)治疗LNCaP异种移植物,平均回归约50%。Mipsagargin (G202)单疗程3天也被观察到对MDA-PCa2b和CWR22R-H具有显著的抗肿瘤作用,持续时间≥30天[2]。 BALB/c小鼠,Mipsagargin (G202) (67 mg/kg;IV)的半衰期为4.9小时[2]。 这与我们之前的报道一致,即MCF-7人类乳腺癌异种移植物在雌激素补充的雌性小鼠中每天静脉注射56 mg/kg/剂量(即40 μmol /kg/剂量)< strong>Mipsagargin (G202)能够产生显著(>50%)的肿瘤消退。当与每日给药口服hdac - 4抑制剂Tasquinimod (TasQ;图7E),有文献证明其能抑制自噬[85]。这些结果保证了每日口服TasQ与间歇性静脉给药Mipsagargin联合的临床试验。 在这些临床前异种移植研究中,Mipsagargin,即使作为单一药物,当其MTD为56 mg/kg/剂量(即40 μmol /kg/剂量)时,也会诱导肿瘤消退。[1] |
| 酶活实验 |
PSMA前药水解[2]
前药用10 μM Fc-PSMA在含0.01 mM CoCl2的tris缓冲液中孵育。在不同时间点去除等分,以0.1%三氟乙酸(TFA)水溶液为溶剂A, 0.1%三氟乙酸(TFA)乙腈为溶剂B,采用高效液相色谱法分析裂解产物,梯度为40% B至100% B,持续25 min。每个水解产物通过共洗脱和质谱鉴定[2]。 钙测量和SERCA泵分析[2] 细胞内游离钙水平的测定采用比例法,用7.5 μM fura-2-乙酰氧基甲酯(分子探针)负载的TSU细胞,如前所述。如前所述,用兔骨骼肌微粒体进行SERCA泵分析。如前所述,用偶联酶法测定SERCA活性作为腺苷5 ' -三磷酸水解速率[2]。 晶体结构测定[2] 兔SERCA1a的12adt - β - asp复合物的增溶和结晶过程,以及晶体稳定和安装过程,基本上与前面对其他TG衍生物的描述相同,详细描述请参见补充材料(方法和表S2)。 |
| 细胞实验 |
细胞毒性检测[2]
为了确定前药的细胞毒活性,我们对psma阴性的TSU人膀胱癌细胞和psma阳性的LNCaP人前列腺癌细胞进行了克隆存活试验。 |
| 动物实验 |
Animal Model: Mice with human breast cancers MCF-7 growing[1]
Dosage: 56 mg/kg Administration: IV; 2 daily; 49 days Result: produced a notable (>50%) tumor regression on its own. When daily dosage of the oral HDAC4 inhibitor Tasquinimod (10 mg/kg/d; oral) was added, this regression stabilized. In vivo studies[2] Maximum tolerated dose (single intravenous dose) was determined and used to perform pharmacokinetic studies. Single-dose pharmacokinetics were assessed by noncompartmental analysis. The AUC from time zero to infinity (AUC0–∞) was calculated with the linear trapezoidal method. The terminal half-life (t1/2) was determined from the terminal slope (Ke) on a log-linear plot of concentration versus time. Biodistribution studies were performed on tumor-bearing mice by harvesting tissues (liver, kidney, skeletal muscle, brain, and tumor) from mice (n = 3) at varying time points after single or multiple intravenous injections of Mipsagargin (G202). (Analytical methods for processing, extracting, and analyzing plasma and tissue drug concentrations are included as supplemental information.)[2] In vivo efficacy studies[2] LNCaP, MDA-PCa2b, or SN12C (2 × 106) cells in 100 μl of Matrigel were inoculated into the flank of 6-week-old male nude mice. MCF-7 cells (2 × 106) were injected into the flanks of female nude mice pretreated with subcutaneous estrogen pellet according to the previously described method. CWR22R-H xenografts were generated by subcutaneous inoculation of 10 mg of minced CWR22R-H tumor in Matrigel as previously described. Statistical analysis of differences in tumor volumes and weights between Mipsagargin (G202) and vehicle controls was performed with Student's t test, and P values of <0.05 were reported in the text. |
| 药代性质 (ADME/PK) |
A phase I trial was initiated to evaluate the safety, tolerability, and PK profile of intravenous (IV) administration of Mipsagargin in patients with locally advanced or metastatic solid tumors, refractory to standard therapy, or for whom no standard therapy was available. These studies established the recommended phase II dosing (RP2D) regimen as IV infusion over a 1hr period of 40 mg m−2 of Mipsagargin on day 1, followed by infusion at the RP2D (66.8 mg/m2 or 1.58 mg/kg) on days 2 and 3 of each 28-day cycle with routine pre-medications and prophylactic hydration on each day of infusion. The dose-limiting toxicities of Mipsagargin included a hypersensitivity reaction that occurred on the first day of the infusion cycle and reversible elevation in serum creatinine that resolved before the end of a treatment cycle. At this infusion dose, Mipsagargin’s estimated half-life of tissue distribution (T1/2-α) is 2 h and its Volume of distribution at steady state (Vss) is 5 L/m2 (i.e., 0.135 L/kg) [80]. This Vss is essentially equivalent to the extracellular fluid volume (i.e., 7.3 L/m2 or 0.2 L/kg) in the human body, consistent with its inability to enter normal tissue. Its elimination phase half-life (T1/2 is 21 h).
|
| 参考文献 |
|
| 其他信息 |
Mipsagargin has been used in trials studying the treatment of Prostate Cancer, Prostatic Neoplasms, Advanced Solid Tumors, Glioblastoma Multiforme, and Hepatocellular Carcinoma, among others.
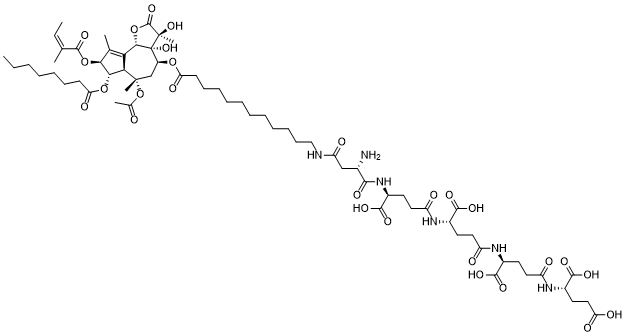
Mipsagargin is a soluble, thapsigargin prodrug containing the cytotoxic analog of thapsigargin, 8-O-(12Aminododecanoyl)-8-O debutanoylthapsigargin (12-ADT) linked, via a carboxyl group, to the targeting peptide containing aspartic acid with potential antineoplastic activity. Upon intravenous administration, mipsagargin targets prostate specific membrane antigen (PSMA), a type II membrane carboxypeptidase, which is overexpressed in prostate cancer cells and in the neovasculature of most solid tumors but not in normal blood vessels. Mipsagargin is subsequently converted, through hydrolysis, into the active cytotoxic analog of thapsigargin 12-ADT-Asp. 12-ADT binds to and blocks the Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase (SERCA) pump, thereby increasing the concentration of cytosolic calcium which leads to an induction of apoptosis. By preventing nutrient supply to tumor cells, G-202 may be able to inhibit tumor growth. Compared to thapsigargin alone, thapsigargin prodrug G-202 is able to achieve higher concentrations of the active agents at the tumor site while avoiding systemic toxicity. |
| 分子式 |
C66H100N6O27
|
|---|---|
| 分子量 |
1409.52
|
| 精确质量 |
1408.66
|
| 元素分析 |
C, 56.24; H, 7.15; N, 5.96; O, 30.65
|
| CAS号 |
1245732-48-2
|
| 相关CAS号 |
1245732-48-2;1627852-87-2 (Trifluoroa cetate);
|
| PubChem CID |
24772106
|
| 序列 |
Asp-{Ggu}-{Ggu}-{Ggu}-{Ggu}
|
| 短序列 |
D{Ggu}-{Ggu}-{Ggu}-{Ggu}
|
| 外观&性状 |
Solid powder
|
| LogP |
-0.4
|
| tPSA |
530Ų
|
| 氢键供体(HBD)数目 |
13
|
| 氢键受体(HBA)数目 |
28
|
| 可旋转键数目(RBC) |
50
|
| 重原子数目 |
99
|
| 分子复杂度/Complexity |
2990
|
| 定义原子立体中心数目 |
13
|
| SMILES |
CCCCCCCC(=O)O[C@H]1[C@H]2C(=C([C@@H]1OC(=O)/C(=C\C)/C)C)[C@H]3[C@]([C@H](C[C@]2(C)OC(=O)C)OC(=O)CCCCCCCCCCCNC(=O)C[C@@H](C(=O)N[C@@H](CCC(=O)N[C@@H](CCC(=O)N[C@@H](CCC(=O)N[C@@H](CCC(=O)O)C(=O)O)C(=O)O)C(=O)O)C(=O)O)N)([C@](C(=O)O3)(C)O)O
|
| InChi Key |
UPYNTAIBQVNPIH-ODMLWHIESA-N
|
| InChi Code |
InChI=1S/C66H100N6O27/c1-8-10-11-17-20-24-51(81)96-55-53-52(37(4)54(55)97-62(91)36(3)9-2)56-66(94,65(7,93)63(92)98-56)44(35-64(53,6)99-38(5)73)95-50(80)23-21-18-15-13-12-14-16-19-22-33-68-48(77)34-39(67)57(82)72-43(61(89)90)27-31-47(76)70-41(59(85)86)25-29-45(74)69-40(58(83)84)26-30-46(75)71-42(60(87)88)28-32-49(78)79/h9,39-44,53-56,93-94H,8,10-35,67H2,1-7H3,(H,68,77)(H,69,74)(H,70,76)(H,71,75)(H,72,82)(H,78,79)(H,83,84)(H,85,86)(H,87,88)(H,89,90)/b36-9-/t39-,40-,41-,42-,43-,44-,53+,54-,55-,56-,64-,65+,66+/m0/s1
|
| 化学名 |
InChI=1S/C66H100N6O27/c1-8-10-11-17-20-24-51(81)96-55-53-52(37(4)54(55)97-62(91)36(3)9-2)56-66(94,65(7,93)63(92)98-56)44(35-64(53,6)99-38(5)73)95-50(80)23-21-18-15-13-12-14-16-19-22-33-68-48(77)34-39(67)57(82)72-43(61(89)90)27-31-47(76)70-41(59(85)86)25-29-45(74)69-40(58(83)84)26-30-46(75)71-42(60(87)88)28-32-49(78)79/h9,39-44,53-56,93-94H,8,10-35,67H2,1-7H3,(H,68,77)(H,69,74)(H,70,76)(H,71,75)(H,72,82)(H,78,79)(H,83,84)(H,85,86)(H,87,88)(H,89,90)/b36-9-/t39-,40-,41-,42-,43-,44-,53+,54-,55-,56-,64-,65+,66+/m0/s1
|
| 别名 |
G-202; G 202; G202; 1245732-48-2; G-202; Mipsagargin [USAN:INN]; UNII-Q032I35QMX; Q032I35QMX; Mipsagargin [INN]; Mipsagargin (USAN/INN); Mipsagargin
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.7095 mL | 3.5473 mL | 7.0946 mL | |
| 5 mM | 0.1419 mL | 0.7095 mL | 1.4189 mL | |
| 10 mM | 0.0709 mL | 0.3547 mL | 0.7095 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT01777594 | Completed | Drug: G-202 | Advanced Adult Hepatocellular Carcinoma |
GenSpera,Inc. | January 2013 | Phase 2 |
| NCT02067156 | Completed | Drug: G-202 | Glioblastoma Multiforme | GenSpera,Inc. | February 2014 | Phase 2 |
| NCT02876003 | Withdrawn | Drug: G-202 | Glioblastoma | GenSpera, Inc. | September 2016 | Phase 2 |
| NCT01056029 | Completed | Drug: G-202 | Advanced Solid Tumors | GenSpera, Inc. | January 2010 | Phase 1 |
| NCT02607553 | Completed | Drug: G-202 | Clear Cell Renal Cell Carcinoma | GenSpera, Inc. | June 2016 | Phase 2 |
| NCT02381236 | Completed | Drug: G-202 | Prostatic Neoplasms | GenSpera, Inc. | February 2016 | Phase 2 |
|
|
|
|
|
|
 DM21-L-G
DM21-L-G
 Mal-VC-PAB-DMEA-PNU-159682
Mal-VC-PAB-DMEA-PNU-159682
 Mc-Pro-PAB-MMAE
Mc-Pro-PAB-MMAE
 Maytansine derivative M24
Maytansine derivative M24
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1



































