| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
Dengue virus (DENV) (EC50 = 0.057 to 11 nM)
|
|---|---|
| 体外研究 (In Vitro) |
登革热是一种主要的健康威胁,四种登革热血清型引起的有症状感染人数估计为9600万1,每年约有1万人死亡2。然而,没有抗病毒药物可用于治疗或预防登革热。研究人员最近将非结构蛋白NS3和NS4B之间的相互作用描述为开发泛血清型登革热病毒(DENV)抑制剂的一个有希望的靶点。在这里,研究人员提出了jnj -1802-一种高效的DENV抑制剂,可以阻断病毒复制复合体内NS3-NS4B的相互作用。JNJ-1802具有皮摩尔到低纳摩尔的体外抗病毒活性[1]。
研究人员利用一种机制病毒动力学模型,根据实验体外感染研究的病毒RNA数据校准,通过表征两种DENV-2菌株在不同JNJ-1802浓度下的感染动力学,评估了JNJ-1802的体外抑制效果。在>1.6 nM的浓度下,DENV-2/RL和DENV-2/16681菌株的最大抑制效果(IC50)分别为1.23x10-02 nM和1.28x10-02 nM,中位浓度为50%。这项工作为JNJ-1802的体外抑制作用提供了重要的见解,并向建模框架迈出了第一步,以支持不同宿主系统中病毒动力学和药物效应的表征。[2] |
| 体内研究 (In Vivo) |
JNJ-1802在体外具有皮摩尔至低纳摩尔的抗病毒活性,对四种DENV血清型中的任何一种感染具有高的抗性屏障和强大的体内功效。最后,我们证明了小分子抑制剂JNJ-1802在非人灵长类动物中对DENV-1或DENV-2病毒感染非常有效。JNJ-1802已成功完成健康志愿者的I期临床研究,并被发现安全且耐受性良好。[2]
首先,研究了JNJ-1802在高剂量DENV-2 (106 PFU;设置1)(图(Fig.2a).2a)。每日两次(b.i.d)给药JNJ-1802 3天导致平均病毒RNA的剂量依赖性减少(与载体相比,剂量组的平均病毒RNA从每毫升3.8 log10到1.0 log10拷贝不等,剂量组的剂量范围为每天60至0.2 mg / kg)。7只小鼠中有4只每天给药60 mg / kg, 14只小鼠中有13只每天给药20 mg / kg,检测不到病毒RNA(图2b)。每天一次(q.d)给药3天(扩展数据图Fig.3a)导致denv感染小鼠的病毒RNA水平低于bid给药,至少对于每公斤30和3 mg给药组(扩展数据图Fig.3b).3b)。此外,每公斤30毫克剂量组的8只小鼠中有5只检测不到病毒RNA水平。相比之下,只有每日给药0.3 mg / kg的剂量比每日给药b.d.d的效果更差(扩展数据图:Fig.3b3b)。[2] 接下来,在一项为期11天的研究(设定2)中,如前所述,评估了JNJ-1802(20,2或0.2 mg / kg / day, b.i.d,持续6天)对AG129小鼠在非致死(102 PFU)病毒攻击后DENV-2复制动力学的影响(扩展数据图Fig. Fig.4a)。给药JNJ-1802对体重没有影响(扩展数据图4b)。我们观察到,在两个最高剂量的8天中,有6天的平均病毒载量与载体相比降低到不可检测的水平(扩展数据图4c,d)。最低剂量未观察到病毒载量显著降低(扩展数据图。图4e4e)。 [2] 综上所述,这些结果通过证明JNJ-1802在小鼠中对所有四种DENV血清型都具有高度活性,扩展了体外数据。 |
| 酶活实验 |
JNJ-1802对DENV-2/16681/eGFP的抗病毒活性是通过表型抗病毒实验确定的,eGFP读数作为病毒复制的衡量标准。同时,使用ATPLite细胞活力发光法测量毒性。实验在参考文献3中描述的三种不同的细胞类型(Vero, Huh-7和THP-1/DC-SIGN)中进行,以排除该化合物的细胞特异性活性。
JNJ-1802对DENV-2 RL株的抗病毒活性在Vero和C6/36细胞中通过RT-qPCR检测病毒RNA水平进行测定。对宿主细胞的潜在毒性作用进行平行测试,同时忽略病毒感染,使用MTS/PMS方法检测Vero细胞,或使用高含量成像方法检测C6/36细胞,如前所述3。JNJ-1802对DENV基因型(补充表1)的抗病毒活性是通过使用RT-qPCR测量病毒RNA来评估的,如前所述3,23。RT-qPCR数据使用QuantStudio 12K Flex软件(v.1.2.3)或SDS v.1.2 Applied Biosystems软件进行分析。使用KaleidaGraph绘图软件绘制抗病毒分子的抑制值。JNJ-1802对DENV-1/45AZ5的抗病毒活性在Vero细胞中通过RT-qPCR检测病毒RNA进行了测试。JNJ-1802对所有不属于黄病毒的其他病毒进行抗病毒活性/毒性试验,使用报告系统、细胞活力、RT-qPCR或斑块减少试验作为读取结果,如补充信息和补充表2中进一步规定。 DENV-2体外抗性选择实验在逐渐增加JNJ-1802浓度的Vero细胞中进行(参考文献3)。如前所述,在每5代和实验结束时(即实验A的第42代和实验B的第50代)对DENV变体进行内部全基因组测序。[2] |
| 细胞实验 |
DENV-2/RL和DENV-2/16681在Vero细胞中的病毒动力学研究[3]
采用两步双工和单工RT-qPCR方法分别测定了JNJ-1802存在和不存在时DENV-2/RL或DENV-2/16681细胞内和细胞外病毒RNA的数量。 下表2给出了每个实验的时间轴概述。简而言之,对于每个菌株和JNJ-1802的每个浓度,在96孔板中以5000个细胞/孔的密度在MEM培养基(2% FBS)中接种16个板。6孔检测JNJ-1802不存在时的病毒动力学,4孔检测JNJ-1802存在时的病毒动力学(浓度范围:0.00256 nM - 8 nM,每个浓度4孔)。对于处理孔,在细胞播种前加入JNJ-1802。接种24 h后,用DENV (MOI 0.01)感染细胞,37℃孵育。16个板中有10个板在实验过程中不刷新培养基,连续10天每天对其中1个板进行进一步处理。对于其他6个培养皿,在感染后第4天更新培养基,从第5天开始,每天收获一个培养皿,直到第10天。因此,对于每个菌株,从总共480个孔中获得病毒RNA测量值。 |
| 动物实验 |
DENV-2 infection studies in mice[2]
Breeding couples of AG129 mice were purchased from Marshall BioResources and bred in-house. The specific pathogen-free status of the mice was regularly checked at the KU Leuven animal facility. Mice were housed in individually ventilated cages (maximum of five mice per cage, type GM500 Sealsafe Plus, Tecniplast) at 21 °C, 55% humidity under a 12 h–12 h light–dark cycle. The mice were provided with food and water ad libitum as well as with cardboard play tunnels and cotton as extra bedding material. Allocation to experimental groups was performed randomly. Housing conditions and experimental procedures were approved by the ethics committee of KU Leuven (licence P169/2011 and P047/2017) following institutional guidelines approved by the Federation of European Laboratory Animal Science Associations. The efficacy of JNJ-1802 on viraemia, viral kinetics and virus-induced disease (survival) was evaluated in DENV-2 infection models in AG129 mice (Supplementary Table 3) as described previously. In brief, in day 3 viraemia studies, DENV female mice (7–11 weeks old) were challenged intraperitoneally (i.p.) with 106 PFU DENV-2 RL strain. Mice were treated b.i.d. by oral gavage for 3 consecutive days with either vehicle (PEG400:water (80:20); n = 24) or various doses of JNJ-1802 (60, 20, 6, 2, 0.6 or 0.2 mg per kg per day; n = 8 (60 mg per kg per day dosing group) and n = 16 (all other dosing groups)), with the first administration 1 h before DENV challenge. In a separate viraemia study, in which mice were orally treated for 3 days with 30, 3 or 0.3 mg per kg per day JNJ-1802 (n = 8 for each group), the treatment was administered once daily. On day 3 after infection, the mice were euthanized, and blood was collected and stored at −80 °C until further use. The effect of the compound on viral RNA levels in the blood on various days after infection was monitored in an in vivo kinetics study. AG129 female mice (aged 7–11 weeks, n = 16 per group) were inoculated i.p. with 102 PFU DENV-2 RL strain. Mice were treated b.i.d. through oral gavage with vehicle or JNJ-1802 using three different doses: 20, 2 or 0.2 mg per kg per day. In the kinetics studies, treatment was initiated 1 h before DENV infection and continued for 6 consecutive days. Each group was subdivided into two smaller groups (A and B; n = 8 each), from which blood was collected on alternating days: on day 1, 3 and 5 (A groups) or on day 2, 4 and 6 (B groups). On day 8 and day 11 after infection, the mice from the A and B groups, respectively, were euthanized, and blood was collected and stored at −80 °C until further use. The survival study was performed as described previously3 with b.i.d. dosing of AG129 mice (aged 7–11 weeks, female, n = 10 per group) at 20, 6, 2 or 0.6 mg per kg per day (start of treatment 1 h before DENV challenge). In delayed-treatment studies (therapeutic setting), AG129 female mice (aged 7–11 weeks, n = 10 per group) were inoculated i.p. with 102 PFU DENV-2 RL strain. Treatment with JNJ-1802 (60 mg per kg per day, b.i.d.) was initiated on day 4 or 5 after infection and continued for 6 days. Mice that were treated with vehicle or JNJ-1802 on the day of infection (that is, 1 h before infection) were included as controls. Each group was subdivided into two smaller groups (A and B; n = 5 per group), from which blood was collected on alternating days: on day 3, 5, 7, and 9 (A groups) or on day 4, 6, 8 and 10 (B groups). On day 12 and day 14 after infection, mice from the A and B groups, respectively, were euthanized, and blood was collected and stored at −80 °C until further use. View More
DENV-1, DENV-3 and DENV-4 infection in mice[2] DENV-2 study in NHPs[2] The animals were selected from the experimental stock from the self-sustainable BPRC colony. Before being placed on the protocol, all animals tested negative for antibodies to DENV serotypes 1, 2, 3 and 4, WNV and ZIKV. All animals were healthy, adult, male and female, Indian-origin rhesus monkeys with a minimum age of 5 years and a minimum body weight of 7 kg. During the experiment, the animals were housed in pairs with a socially compatible cage mate under biosafety level 3 (BSL3) conditions. The animals were offered a daily diet consisting of monkey food pellets, fruit and bread. Enrichment (toys, extra food) was offered daily. Drinking water was available ad libitum through an automatic watering system. The animals were randomly allocated to treatment groups according to a randomized block design on the basis of the sex and body weight of the animals. To assess the efficacy of JNJ-1802 in a prophylactic setting, rhesus macaques received daily vehicle (100% PEG400, n = 3), or compound JNJ-1802 at a dose of 0.01 mg per kg per day (n = 3), 0.18 mg per kg per day (n = 3) or 3 mg per kg per day (n = 3) dissolved in 100% PEG400 through oral gavage, starting from 1 day before experimental infection until 10 days after infection. On day 0, the animals were exposed through intradermal inoculation in the upper back with 100 TCID50 of strain DENV-2/16681. In the DENV-1 NHP study that was conducted first, infection with DENV-1 was performed 3 days after oral administration of JNJ-1802. As a rapid increase in drug levels was obtained 12 h after dosing and high concentration persisted up to the end of the dosing period, animals in the DENV-2 NHP study were infected already one day after the start of oral administration with JNJ-1802. The animals were monitored daily during the study period for general behaviour until day 28. Blood samples were taken at regular timepoints for both virological assessments and determination of plasma compound concentrations. Virological assessments included the quantification of DENV-2 RNA using RT–qPCR, the detection of DENV NS1 antigen by ELISA, the quantification of infectious DENV-2 by TCID50 assay in Vero cells and the detection of anti-DENV IgM/IgG by ELISA. Details of the assessment of haematological parameters are provided in the Supplementary Information. Pharmacokinetics studies[2] The pharmacokinetics profile of JNJ-1802 was evaluated in a separate experiment in fed male CD-1 mice (n = 3 per group, 27–33 g body weight, aged around 6–8 weeks; Charles River Laboratories) at Janssen Pharmaceutica NV. The housing conditions and experimental procedures were approved by the ethics committee on animal experiments of Janssen Pharmaceutica NV (license number LA1100119). Janssen Pharmaceutica NV has full AAALAC accreditation. The mice were intravenously injected into the tail vein with 2.5 mg per kg of JNJ-1802, which was formulated as an 0.5 mg ml−1 solution in 70% PEG400/30% H2O, and blood samples were collected (in EDTA-containing microcentrifuge tubes) from the saphenous vein at 0.12, 0.33, 1, 2, 4, 7 and 24 h after dosing. Moreover, JNJ-1802 was administered by oral gavage at 1, 3, 10 and 30 mg per kg, formulated as a solution in 80% PEG400/20% H2O, and the blood samples were collected from the saphenous vein at 0.5, 1, 2, 4, 7 and 24 h after dosing. The blood samples were immediately centrifuged at 4 °C and the plasma was stored at −20 °C. Compound concentrations in the plasma samples were determined using the API 5500 LC–MS/MS system mass spectrometer (Applied Biosystems). Individual plasma concentration–time profiles were processed for a non-compartmental pharmacokinetics analysis using Phoenix WinNonlin v.6.3. [2] The pharmacokinetics profile of JNJ-1802 was evaluated in female monkeys after single intravenous administration and in DENV-infected monkeys after repeated oral administration. After single intravenous administration, the monkey blood was collected at the BPRC just before dosing, and at 0.05, 0.15, 0.5, 4, 6, 8, 24, 48 and 264 h after dosing. After oral administration, the monkey blood was collected at the BPRC on the first and last day of dosing just before dosing and at 1, 2, 4, 6, 8 and 24 h after dosing that day. Moreover, blood samples were taken on days 3, 6 and 7 after infection just before dosing. For the study at WRAIR, the monkey blood was collected just before dosing and at 1, 4, 8 and 24 h after the first day of dosing and before dosing and at 2, 5, 8, 24 and 96 h after the last day of dosing. Furthermore, blood samples were taken on days 0, 2, 5 and 8 after infection just before dosing. Blood was drawn from the femoral vein (or a peripheral vein, for example, saphenous or cephalic) The volume of blood collected did not exceed 7.5% of an animal’s total blood volume over a 7 day period or 10% over a 2 week period. A volume of 1 ml blood was collected into a blood collection tube coated with EDTA as anti-coagulant. Within 1 h after collection, the blood samples were centrifuged in a precooled centrifuge for 10 min at 1,500g (4 °C). Within 2 h after blood collection, the plasma samples were stored at −80 °C until shipment. Individual plasma concentration–time profiles were processed for a non-compartmental pharmacokinetics analysis using Phoenix WinNonlin v.6.3. |
| 药代性质 (ADME/PK) |
JNJ-1802 has a favourable pharmacokinetic profile in mice. The compound has low clearance and a moderate volume of distribution resulting in a terminal half-life of 6.2 h. The plasma protein binding in mice was higher than 99.9%. The oral bioavailability was 46% and 59% at 1 and 3 mg per kg, respectively, and increased to more than 100% due to more than dose-proportional pharmacokinetics between 3 and 10 mg per kg. In a separate study, after single oral dosing, JNJ-1802 showed exposures in mice with an area under the plasma concentration versus time curve during 24 h (AUC0–24h) ranging from 1,520 up to 120,000 ng h ml−1 for a dose ranging from 1 mg per kg to 30 mg per kg, respectively (Extended Data Table 2a,b).[2]
|
| 参考文献 |
|
| 其他信息 |
Background: Dengue is a growing global health threat with no specific antiviral drugs available for treatment or prophylaxis. This first-in-human, double-blind, randomized, placebo-controlled study aimed to examine the safety, tolerability, and pharmacokinetics of increasing single and multiple oral doses of JNJ-1802, a pan-serotype dengue antiviral small molecule.
Methods: Eligible healthy participants (18-55 years of age) were randomized to receive oral JNJ-1802 in fasted conditions as (1) single doses (50-1200 mg; n = 29) or placebo (n = 10); or (2) once-daily doses (50-560 mg for 10 consecutive days or 400 mg for 31 days; n = 38) or placebo (n = 9). Safety and tolerability were evaluated throughout the study. Plasma and urine samples were collected at predetermined time points to characterize pharmacokinetics.
Results: JNJ-1802 was generally safe and well-tolerated. One grade 3 adverse event (depression) was reported but not considered drug-related by the investigator. Two grade 2 events of rash occurred (multiple-dose part) that were considered very likely related to JNJ-1802 by the investigator and resolved. No clinically relevant changes were observed in laboratory tests, electrocardiograms, or vital signs.JNJ-1802 exposure after single or multiple doses increased dose-proportionally from 50 to 150 mg and less than dose-proportionally for higher doses. The terminal elimination half-life was 6.3-9.2 days and the accumulation factor was 4.3-7.3 after 10 days and 14.6 after 31 days with low amounts of unchanged drug in urine (<0.001% of the 400 mg dose).
Conclusions: Pharmacokinetics and safety results of JNJ-1802 support further clinical development for the treatment and prevention of dengue infection.[1]
Dengue is a major health threat and the number of symptomatic infections caused by the four dengue serotypes is estimated to be 96 million1 with annually around 10,000 deaths2. However, no antiviral drugs are available for the treatment or prophylaxis of dengue. We recently described the interaction between non-structural proteins NS3 and NS4B as a promising target for the development of pan-serotype dengue virus (DENV) inhibitors3. Here we present JNJ-1802-a highly potent DENV inhibitor that blocks the NS3-NS4B interaction within the viral replication complex. JNJ-1802 exerts picomolar to low nanomolar in vitro antiviral activity, a high barrier to resistance and potent in vivo efficacy in mice against infection with any of the four DENV serotypes. Finally, we demonstrate that the small-molecule inhibitor JNJ-1802 is highly effective against viral infection with DENV-1 or DENV-2 in non-human primates. JNJ-1802 has successfully completed a phase I first-in-human clinical study in healthy volunteers and was found to be safe and well tolerated4. These findings support the further clinical development of JNJ-1802, a first-in-class antiviral agent against dengue, which is now progressing in clinical studies for the prevention and treatment of dengue.[2] |
| 分子式 |
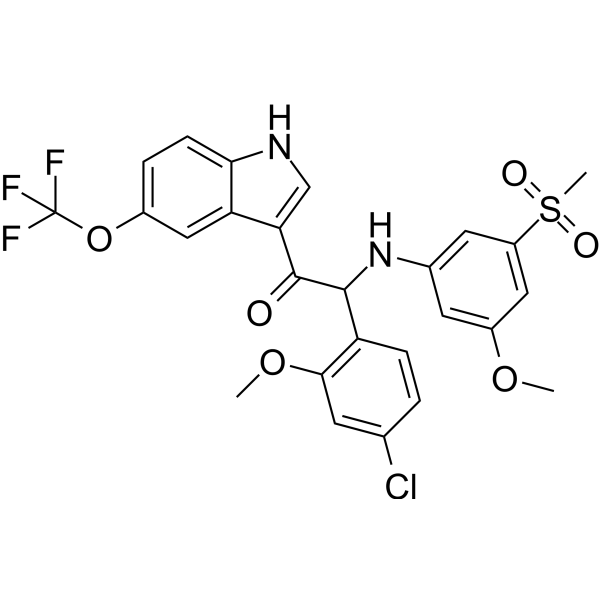
C26H22CLF3N2O6S
|
|---|---|
| 分子量 |
582.98
|
| 精确质量 |
582.08391
|
| CAS号 |
2890688-86-3
|
| PubChem CID |
133083036
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
6.1
|
| tPSA |
115Ų
|
| InChi Key |
QNOPDDHSGQQLCV-DEOSSOPVSA-N
|
| InChi Code |
InChI=1S/C26H22ClF3N2O6S/c1-36-17-9-15(10-18(11-17)39(3,34)35)32-24(19-6-4-14(27)8-23(19)37-2)25(33)21-13-31-22-7-5-16(12-20(21)22)38-26(28,29)30/h4-13,24,31-32H,1-3H3/t24-/m0/s1
|
| 化学名 |
(2S)-2-(4-chloro-2-methoxyphenyl)-2-(3-methoxy-5-methylsulfonylanilino)-1-[5-(trifluoromethoxy)-1H-indol-3-yl]ethanone
|
| 别名 |
5EJO0RJF9O; 2-(4-Chloro-2-methoxyphenyl)-2-((3-methoxy-5-(methylsulfonyl)phenyl)amino)-1-(5-(trifluoromethoxy)-1H-indol-3-yl)ethanone, (S)-; (+)-(2S)-2-(4-Chloro-2-methoxyphenyl)-2-((3-methoxy-5-(methylsulfonyl)phenyl)amino)-1-(5-(trifluoromethoxy)-1H-indol-3-yl)ethanone; Ethanone, 2-(4-chloro-2-methoxyphenyl)-2-((3-methoxy-5-(methylsulfonyl)phenyl)amino)-1-(5-(trifluoromethoxy)-1H-indol-3-yl)-, (+)-; (S)-Mosnodenvir; mosnodenvir [INN]; UNII-5EJO0RJF9O;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
|---|
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7153 mL | 8.5766 mL | 17.1532 mL | |
| 5 mM | 0.3431 mL | 1.7153 mL | 3.4306 mL | |
| 10 mM | 0.1715 mL | 0.8577 mL | 1.7153 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 SARS-CoV-2 main protease
SARS-CoV-2 main protease
 AH-D peptide TFA
AH-D peptide TFA
 AG-1859
AG-1859
 NS2B/NS3-IN-9
NS2B/NS3-IN-9
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
