| 规格 | 价格 | |
|---|---|---|
| 5mg | ||
| Other Sizes |

| 靶点 |
Plasmepsin IX/X (PMIX/X); Plasmodium
|
|---|---|
| 体外研究 (In Vitro) |
WM382 抑制恶性疟原虫和间日疟原虫,对恶性疟原虫的 IC50 值为 0.6 nM,并对 HepG2 细胞表现出相当大的细胞毒性 (IC50=24.8 μM)[2][3]。 WM382 的 Ki 值分别为 13.4 μM 和 0.035 nM,选择性结合 PMV 和 PMX[3]。在伯氏疟原虫感染的 HepG2 体外培养物中,WM382(1 nM 和 100 nM)提醒注射后达到血液感染的时间[3]。
|
| 体内研究 (In Vivo) |
在伯氏疟原虫和恶性疟原虫寄生虫小鼠模型中,WM382(20 mg/kg,每日两次或 1-30 mg/kg,每日一次;口服;持续 4 天)可有效消除感染。 WM382 可以有效治疗人源化小鼠的无性恶性疟原虫感染,同时还能阻止蚊子传播[3]。
|
| 酶活实验 |
恶性疟原虫白酶加工抑制试验[2]
蛋 加工抑制试验基本上按照之前的描述进行(Favuzza等人,2020)。将蛋白酶抑制剂添加到同步的后期滋养体/早期分裂体培养物中,然后通过LD或LS磁柱去除未感染的红细胞。WM4和WM382分别以40 nM和2.5 nM的终浓度加入。不含蛋白酶抑制剂的对照盘。用含有相同浓度抑制剂的完整RPMI 1640培养基洗脱柱上的寄生虫。将洗脱的寄生虫调整为5 × 106个/mL,每孔添加150 mL, 96孔平底培养皿。寄生虫培养16小时,并涂片代表性孔进行吉氏染色,以确保破裂正常发生(对照孔)或破裂被阻断(WM4, WM382>条件)。寄生虫以10000 g/10 min离心,收集分殖子和上清。将两个部分的蛋白质加入还原样品缓冲液中,用4-12%或3-8%的丙烯酰胺凝胶分离,用于后续的免疫印迹。 表面等离子体共振[2] PvPMX或PfPMX蛋白酶通过胺偶联固定在CM5传感器芯片上,在10 mM激活的pH 5.5中,以10 mL min - 1的流速注射420 s,通常固定约4000 RU的蛋白质。所有实验均在HBS-EP缓冲液(10 mM Hepes pH 7.4, 150 mM NaCl, 3 mM EDTA和0.05%吐温20)中进行,温度为18°C,流速为30 mL min - 1。将化合物WM4和WM382在HBS-EP +缓冲液中稀释至0.031 nM - 16 nM范围内的适当浓度。接触时间为90 s,分离时间为1500 s,然后用50 mM甘氨酸pH 9.5进行30 s再生。使用新固定的传感器表面独立进行了三次实验。PvPMX数据采集于BIAcore S200仪器,数据分析软件版本为BIAcore S200 1.1; PfPMX数据采集于BIAcore 8K+仪器,数据分析软件版本为BIAcore Insight 3.0.12.15655。所有传感器图均采用活化的乙醇胺阻断传感器表面和HBS-EP +缓冲空白进行双重参考。使用1:1的结合模型拟合WM4的打开和关闭率来确定亲和力,由于关闭率太慢,无法报告WM382的亲和力。 CETSA热稳定性测定[3] Lysate CETSA实验基本上按照描述进行(Dziekan et al., 2019)。CETSA研究样品采用高度同步分裂期恶性疟原虫3D7-PMIX_HA和3D7-PMX_HA寄生虫制备。后期寄生虫(入侵后40-44小时)通过Percoll密度梯度纯化,并与1 μM化合物1 (C1)抑制剂(内部合成)孵育(Hale等,2017)。孵育2-4 h后,成熟分裂体用PBS洗涤1次,感染的红细胞在20体积0.15% (w/v)皂苷的PBS中裂解,冰孵育20分钟。寄生虫在冰冷的PBS中洗涤3次,最终的球在10体积裂解缓冲液(0.4% NP-40 (Roche) / PBS)中重悬,通过3次冷冻(干冰/乙醇浴)-解冻循环裂解。裂解物在16000 g离心30分钟后清除,含有可溶性寄生虫蛋白的上清液在−80℃保存至使用。将化合物601 (5 μM)、WM4 (2 μM)和WM382 (1 μM)加入到8 × 50 μg蛋白裂解液等分液中(蛋白质浓度通过BCA蛋白测定法测定),室温(RT)孵育3min,在Biorad T100热循环器中分别在65-40℃温度梯度下加热3min,然后在4℃孵育3min。加热后的裂解物在4℃下13000 g离心30分钟。根据制造商的说明,在预制的4%-12%梯度凝胶上用MES运行缓冲液溶解可溶性蛋白质。 |
| 细胞实验 |
敲除疟原虫的EC50测定[3]
培养环期恶性疟原虫3D7、3D7- pmix - ha和3D7- pmix - ha,同时增加GlcN (Sigma)浓度。37℃孵育72 h后,用0.06%皂苷溶解滋养体感染的红细胞,用2倍还原SDS-PAGE样品缓冲液溶解微球,进行抗ha免疫检测。如前面所述,在2.5 nM GlcN(分别是ha标记蛋白的正常表达和减少表达)缺失和存在的情况下,通过FACS和SYBR Green测定WM4和WM382对恶性疟原虫3D7、3D7- pmix - ha和3D7- pmx - ha寄生虫的抑制EC50。 药物杀伤时间[3] 用5%山梨醇(Sigma)同步3D7寄生虫培养,每隔46 h同步两次,然后当培养中混合了晚分裂体和早分裂体时再次同步。在3%环数和4%红细胞压积的条件下,分别建立3份10ml的培养液,分别含有80nM WM4、40nM WM5、5nM WM382或DMSO (Sigma)载体对照。每个培养物每8 h定量一次,共48 h,采集50 μL样品,流式细胞术计数(如前所述)。用吉姆萨染色血膜显微镜检查各时间点寄生虫的发育阶段。在32 h时间点更换培养基和化合物。 入侵检测[3] 山梨醇处理与环期添加WM4 (40 nM)、WM382 (2.5 nM)或DMSO对照同步培养3D7寄生虫。后期寄生体(入侵后40 h)通过磁分离富集,发育为完全节段的分裂体寄生体。为了防止分裂体破裂和分裂子释放,对照寄生虫用1 μM化合物1 (C1)孵育。孵育5-6小时后,将寄生液泡膜封闭的分生子(PEMS)制成颗粒,在小体积完整培养基(含WM4, WM382或C1)中重悬,并通过1.2 μm注射器过滤器(Acrodisc;32毫米;棺罩)。将含有纯化的分裂子的滤液立即加入新鲜红细胞(70%-80%的红细胞比容)中,在摇床(1100转/分)中37℃孵育20分钟,使宿主细胞浸润,然后稀释至2%的红细胞比容。37℃孵育24小时后,通过显微镜(giemsa染色薄涂片)和流式细胞术测量寄生虫率来评估侵袭情况 电阻时间的测定[3] 实验1将105、106、107、108和109株Dd2寄生虫分别暴露于10 nM的atovaquone或1.5 nM WM382环境中,监测90 d。实验2将106、107和108 Dd2寄生虫的三次培养分别暴露于5 nM的atovaquone、80 nM的WM5或1.5 nM WM382中,监测62天。每个实验都通过每周一次的血膜显微镜检查来监测。每周更换三次培养基和化合物。 |
| 动物实验 |
动物/疾病模型:感染伯氏疟原虫的小鼠[3]
剂量:20 mg/kg 给药途径:灌胃(po);每日两次,持续4天;监测30天 实验结果:治愈了伯氏疟原虫感染的小鼠,并预防了肝脏引起的血液感染。 动物/疾病模型:人源化非肥胖糖尿病-重症联合免疫缺陷(NOD-scid)IL2Rgnull小鼠模型(NSG)[3] 剂量:1、3、10、30 mg/kg 给药途径:灌胃(po);每日一次,持续4天;监测7天 实验结果: 30 mg/kg剂量组在第6天或3 mg/kg和10 mg/kg剂量组在第7天清除寄生虫血症。 伯氏疟原虫感染小鼠剂量范围试验[3] 在剂量范围研究中,小鼠分别以每日两次(bid)给药方案(剂量分别为30、10、3或1 mg/kg/天,图4B)或每日一次(qd)给药方案(剂量分别为60、30或10 mg/kg/天,图4C)口服WM382,连续4天,首次给药时间为感染后2小时。对照组小鼠以每日一次(qd)给药方案(剂量为10 mg/kg/天)口服氯喹,连续4天。感染后第2天至第30天,每日采用流式细胞术和显微镜检查法检测寄生虫血症,如上所述。感染后第30天存活且外周血中未检测到寄生虫的动物被认为已治愈(即100%疗效)。 恶性疟原虫人源化NOD-scid IL2R_null小鼠模型[3] 在小鼠恶性疟原虫SCID模型中测试化合物(Angulo-Barturen等人,2008)。简而言之,将WM382配制于20% DMSO、60%丙二醇和20%水中,并给予一组年龄匹配的、先前已移植人红细胞的免疫缺陷NOD-scid IL-2Rγnull雌性小鼠。小鼠经静脉注射感染2 × 10⁷个恶性疟原虫Pf3D7感染的红细胞(第0天)(Angulo-Barturen等,2008)。感染后第3天,将小鼠随机分配至不同治疗组,连续4天每天一次,通过灌胃给予10 mL/kg的药物(每组n = 3只小鼠)。采用显微镜检查法测定寄生虫血症。使用抗鼠红细胞 TER119 单克隆抗体和 SYTO-16 通过流式细胞术监测嵌合体,然后对每 24 小时采集的 2 μL 连续血样进行流式细胞术分析,直至实验完成。 伯氏疟原虫肝脏寄生虫生长及向血液感染转变的体内分析 [3] 雌性 BALB/c 小鼠通过静脉注射或感染性蚊虫叮咬感染 PbmCherryLuci 子孢子。小鼠(通过任一途径感染)未接受任何治疗或接受 WM382(制备方法如前所述)口服治疗,剂量和感染后时间 (hpi) 见图示。对于静脉注射,将新鲜分离的唾液腺子孢子用玻璃棉过滤以去除杂质,然后在注射前将40,000个子孢子重悬于200 μL施耐德昆虫培养基中。对于蚊虫叮咬感染,根据蚊子携带卵囊的百分比,将5只感染的蚊子放入单独的喂食杯中(例如,如果该批次蚊子中有83%携带卵囊,则每个喂食杯中放入6只蚊子)。用氯胺酮/甲苯噻嗪麻醉小鼠,然后将其放置在单独的喂食杯中开始感染。允许蚊子叮咬小鼠15分钟,每分钟将小鼠在不同的喂食杯之间轮换,以促进蚊子探查并确保所有小鼠均等地暴露于感染性蚊虫叮咬。从感染后第3天到第30天,每天监测小鼠吉姆萨染色血涂片中是否存在寄生虫,如果在此期间血涂片检测结果始终为阴性,则认为小鼠已免受血液感染。 Peters 小鼠伯氏疟原虫感染4天抑制试验[3] 对于图1所示的结果,将1 × 10⁶个从先前感染的供体小鼠体内抽取的、被伯氏疟原虫ANKA株寄生的红细胞腹腔注射(IP)给雄性瑞士小鼠。测试化合物配制于由10% DMSO/90% Solutol(5% Solutol® HS-15溶于0.9%生理盐水)组成的溶剂中。感染后2小时,小鼠连续4天(每日一次,qd方案)接受腹腔注射试验化合物(WM4、WM5:20 mg/kg)或氯喹(10 mg/kg),或接受腹腔注射溶剂作为对照。末次给药后24小时采集外周血样本,并通过吉姆萨染色血涂片的显微镜分析检测寄生虫血症。寄生虫血症数值为每组6只小鼠的平均值,并以寄生虫血症百分比表示。如图 4A 所示,将表达 GFP 的血期伯氏疟原虫(P. berghei ANKA GFPcon 259cl2 (Franke-Fayard et al., 2004))腹腔注射(IP)给“供体”雌性瑞士小鼠。三天后,将 4 只一组的“受体”瑞士小鼠静脉注射(IV)来自“供体”小鼠的 1 × 10⁷ 个感染红细胞。感染两小时后,实验小鼠(每组 4 只)不进行任何处理(对照组),或连续 4 天口服测试药物(溶于 20% DMSO/60% PG/20% 水 (v/v/v) 溶液)或氯喹(溶于水)。小鼠以每日两次(bid)给药方案接受 WM382 治疗,持续 4 天。每日给予小鼠 mpk,首次给药时间为感染后 2 小时。末次给药后 12 小时采集外周血样本,采用流式细胞术(使用 BD 公司的 FACSCalibur 流式细胞仪,检测 100,000 个记录事件中 GFP 阳性细胞的比例)和吉姆萨染色血涂片显微镜分析法测定寄生虫血症。每组 4 只小鼠的寄生虫血症数值为平均值,以寄生虫血症百分比表示。 |
| 药代性质 (ADME/PK) |
口服生物利用度(大鼠为 8%,小鼠为 38%)
|
| 毒性/毒理 (Toxicokinetics/TK) |
血浆蛋白结合率 = 95%
|
| 参考文献 |
[1]. Manuel de LR, et al. The Invention of WM382, a Highly Potent PMIX/X Dual Inhibitor toward the Treatment of Malaria. ACS Med Chem Lett. 2022 Oct 12.
[2]. Hodder AN, et al. Basis for drug selectivity of plasmepsin IX and X inhibition in Plasmodium falciparum and vivax. Structure. 2022 Jul 7;30(7):947-961.e6. [3]. Favuzza P, et al. Dual Plasmepsin-Targeting Antimalarial Agents Disrupt Multiple Stages of the Malaria Parasite Life Cycle. Cell Host Microbe. 2020 Apr 8;27(4):642-658.e12. |
| 其他信息 |
对包括青蒿素在内的一线抗疟药物的耐药性日益增强,因此迫切需要发现具有全新作用机制的新药。在沃尔特和伊丽莎·霍尔医学研究所的合作以及惠康基金会的资助下,默克公司利用其天冬氨酸蛋白酶抑制剂库进行表型筛选,发现了一系列比氯喹更有效的疟原虫蛋白酶X (PMX) 抑制剂。受PMX同源模型的启发,研究人员致力于优化这些抑制剂的效力,最终发现了一些先导化合物,这些化合物不仅能有效抑制PMX,还能抑制另一种重要的天冬氨酸蛋白酶——疟原虫蛋白酶IX (PMIX)。进一步的效力和药代动力学优化工作最终发现了WM382,这是一种非常有效的PMIX/X双重抑制剂,在疟原虫生命周期的多个阶段均具有强大的体内疗效,且耐药性极佳。[1]
疟原虫蛋白酶IX (PMIX) 和X (PMX) 是疟原虫属寄生虫出胞、入侵和发育所必需的天冬氨酸蛋白酶。WM4和WM382均能抑制恶性疟原虫和间日疟原虫中的PMIX和PMX。WM4抑制PMX,而WM382是PMIX和PMX的双重抑制剂。为了解它们的功能,我们鉴定了它们的蛋白质底物。酶动力学和结构分析揭示了决定药物特异性的相互作用。PMIX和PMX具有相似的底物特异性;然而,它们对肽类和蛋白质底物的特异性存在显著差异。 WM4 和 WM382 与 PMIX 和 PMX 结合的差异主要体现在 S' 区的结构变异以及活性位点 S3 口袋的参与情况上。PMX 的结构揭示了药物结合的相互作用和机制细节,这对于开发针对这些靶点的临床候选药物至关重要。[2] 青蒿素联合疗法 (ACT) 是治疗疟疾的主要方案,疟疾是由胞内寄生虫疟原虫引起的。然而,ACT 耐药性的增加凸显了寻找新药的重要性。最近,天冬氨酸蛋白酶疟原虫蛋白酶 IX 和 X (PMIX 和 PMX) 被认为是很有前景的药物靶点。在本研究中,我们描述了 PMIX 和 PMX 的双重抑制剂,包括 WM382,它们可以阻断疟原虫生命周期的多个阶段。我们证明 PMX 是裂殖子入侵的关键调节因子,并直接调控入侵、寄生虫发育和释放所需的蛋白质的成熟。口服 WM382 可以治愈小鼠的伯氏疟原虫感染,并预防肝脏引起的血液感染。此外,WM382 对人源化小鼠体内的恶性疟原虫无性感染有效,并能阻止其传播给蚊子。体外培养无法筛选出耐药的恶性疟原虫。综上所述,这些结果表明,PMIX 和 PMX 双重抑制剂是治疗和预防疟疾的有希望的候选药物。[3] |
| 分子式 |
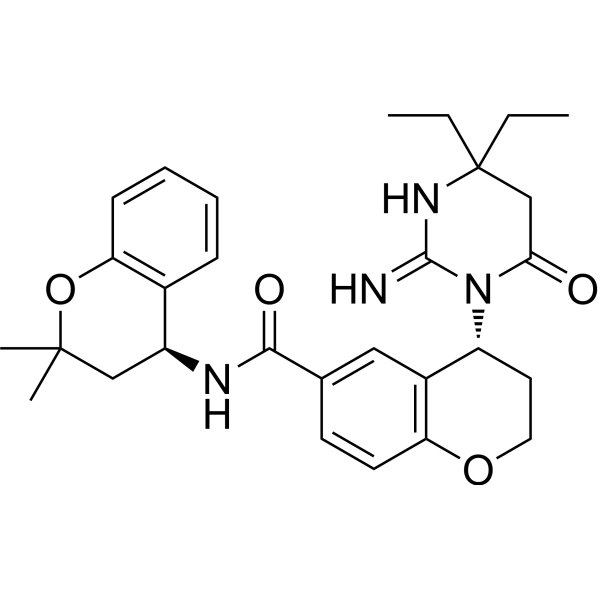
C29H36N4O4
|
|---|---|
| 分子量 |
504.620547294617
|
| 精确质量 |
504.273
|
| 元素分析 |
C, 69.02; H, 7.19; N, 11.10; O, 12.68
|
| CAS号 |
2606990-92-3
|
| PubChem CID |
154699453
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
3.4
|
| tPSA |
106
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
37
|
| 分子复杂度/Complexity |
900
|
| 定义原子立体中心数目 |
2
|
| SMILES |
CCC1(CC(=O)N(C(=N1)N)[C@@H]2CCOC3=C2C=C(C=C3)C(=O)N[C@H]4CC(OC5=CC=CC=C45)(C)C)CC
|
| InChi Key |
ZSZSSSHEMYULPX-FCHUYYIVSA-N
|
| InChi Code |
InChI=1S/C29H36N4O4/c1-5-29(6-2)17-25(34)33(27(30)32-29)22-13-14-36-23-12-11-18(15-20(22)23)26(35)31-21-16-28(3,4)37-24-10-8-7-9-19(21)24/h7-12,15,21-22H,5-6,13-14,16-17H2,1-4H3,(H2,30,32)(H,31,35)/t21-,22+/m0/s1
|
| 化学名 |
(4R)-4-(2-amino-4,4-diethyl-6-oxo-5H-pyrimidin-1-yl)-N-[(4S)-2,2-dimethyl-3,4-dihydrochromen-4-yl]-3,4-dihydro-2H-chromene-6-carboxamide
|
| 别名 |
WM-382; WM382; 2606990-92-3; CHEMBL5171401; (4R)-4-(2-amino-4,4-diethyl-6-oxo-5H-pyrimidin-1-yl)-N-[(4S)-2,2-dimethyl-3,4-dihydrochromen-4-yl]-3,4-dihydro-2H-chromene-6-carboxamide; (4R)-4-[(2E)-4,4-diethyl-2-imino-6-oxo-1,3-diazinan-1-yl]-N-[(4S)-2,2-dimethyl-3,4-dihydro-2H-1-benzopyran-4-yl]-3,4-dihydro-2H-1-benzopyran-6-carboxamide; I0L; SCHEMBL22997111; GTPL11162; WM 382
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : 5 mg/mL (9.91 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9817 mL | 9.9084 mL | 19.8169 mL | |
| 5 mM | 0.3963 mL | 1.9817 mL | 3.9634 mL | |
| 10 mM | 0.1982 mL | 0.9908 mL | 1.9817 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
