| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
SETD2 (IC50=18 nM)
|
|---|---|
| 体外研究 (In Vitro) |
EZM0414 抑制一组 MM 和 DLBCL 细胞系,t(4;14) 细胞的 IC50 为 0.24 μM,DLBCL 细胞系的 IC50 为 0.023 μM->10 μM [2]。
EZM0414对SETD2的抑制在MM和DLBCL细胞系中产生了有效的抗增殖作用。EZM0414对t(4;14)和非t(4;14) MM细胞系的增殖均有抑制作用,在MM细胞系的t(4;14)亚群中普遍观察到较高的抑制增殖活性。EZM0414在t(4;14)细胞系中的IC 50中值为0.24 μM,而在非t(4;14)细胞系中为1.2 μM。此外,对DLBCL细胞株的生长抑制作用表现出广泛的敏感性,ic50值从0.023 μM到bbb10 μM。[2] 在由47个靶点和72个激酶组成的安全面板中,EZM0414的体外测试显示,除D2 (IC50 = 13.0 μM,拮抗剂)和5-HT1B (IC50 = 3.2 μM,激动剂)外,所有靶点的IC50都在25 μM左右。[3] |
| 体内研究 (In Vivo) |
在植入人 KMS-11 细胞的 NOD SCID 小鼠异种移植模型中,EZM0414(15 和 30 mg/kg,口服,BID,每日)可抑制肿瘤生长,且耐受性良好 [3]。在大鼠和小鼠中,EZM0414(50 mg/kg,口服)的口服生物利用度接近 100%,小鼠的 t1/2 值为 1.8 小时,大鼠为 3.8 小时 [3]。
在3个MM和4个DLBCL细胞系来源的异种移植模型中,与对照相比,EZM0414具有统计学上显著的抗肿瘤活性。在t(4;14) MM细胞系衍生的异种移植物模型KMS-11中,在前两个剂量下观察到强劲的肿瘤生长回归,最大TGI为95%。此外,两个非t(4;14) MM (RPMI-8226, MM. 1s)和两个DLBCL异种移植模型(TMD8, KARPAS422)显示> 75% TGI;另外两种DLBCL模型(WSU-DLCL2, SU-DHL-10)对EZM0414的反应显示出50%的TGI。在所有测试的模型中,观察到的抗肿瘤作用与肿瘤内H3K36me3水平的降低相关,表明体内SETD2甲基转移酶活性的靶向抑制。[2] |
| 酶活实验 |
SETD2(1434-1711)测定[3]
生化试验监测了s -腺苷-蛋氨酸(SAM)的氚化甲基与残基26-40对应的生物素化组蛋白3肽的结合。底物肽的序列为biotin- ahx - rksapatggvkkkphr - nh2和3H-SAM,购自American radiollabelled Chemicals, Inc.。在384孔分析板上,将40L酶与1L化合物或DMSO孵育30分钟,然后与10L底物溶液开始反应。实验在室温下进行,实验缓冲液由25 mM比辛,pH 8.0, 7.5 mM-巯基乙醇,0.002%吐温-20和0.01%牛皮肤明胶(BSG)组成。在产物形成的线性部分,用10L的1 mM s -腺苷-同型半胱氨酸(SAH)和1 mM SAM来淬灭反应。从淬火反应中,将50L转移到链霉亲和素涂覆的闪光板 上,孵育至少2h,然后用0.1% Tween-20洗涤一次。链霉亲和素包被板捕获的3h标记肽的信号通过Topcount板读取器计数。抑制百分比(%I)和IC50值分别由式1和式2计算。%𝐼=(1−(𝑆−𝑚𝑖𝑛𝑚𝑎𝑥−𝑚𝑖𝑛))(eq - 1) %𝐼=(100−𝑏𝑜𝑡𝑡𝑜𝑚)(1 +(𝐼𝐶50𝐼)𝑛)+𝑏𝑜𝑡𝑡𝑜𝑚(eq 2)分钟的信号完全抑制SETD2从井20嗯SAH的最终浓度和最大的信号从井DMSO而不是化合物。计算IC50时,bottom为理论最小%I, I为缓蚀剂浓度,n为Hill斜率。化合物IC50的测定是通过测试10个浓度的化合物,将其稀释3倍,最终浓度为4 nM酶和底物浓度等于0.7M肽和2M SAM的KM值。 筛选方法:[3] 384孔源板中的EZM0414 (10 nL)直接加入到包被聚d -赖氨酸(PDL)的384孔培养板中。将A549细胞接种于浓度为80000细胞/ mL的实验培养基中,并添加到PDL包被板上,每孔体积为50µL。将培养皿放在实验台上约20分钟,使细胞在孔底沉淀。37℃,5% CO2孵育3天。三天后,将培养皿从培养箱中取出,让培养皿达到室温。将培养基从板中吸出,每孔加50µL的100%甲醇,孵育30分钟。用抽吸法去除甲醇,每孔115µL洗涤缓冲液(1X PBS加0.05%吐温-20 (v/v))洗涤3次。接下来,每孔加入50µL奥德赛阻断缓冲液+ 0.1%吐温-20,在室温下孵育1小时。去除阻断缓冲液,洗涤3次,每孔加入20µL一抗(抗组蛋白H3三甲基K36在奥德赛缓冲液中以0.1%吐温-20 (v/v)稀释1:1000),在4°C下孵育过夜(16小时)。每孔115µL洗涤缓冲液(1X PBS加0.05% Tween-20 (v/v))洗涤5次。每孔加入20µL二抗(1:500 800CW山羊抗兔IgG (H+L)抗体,1:1000 DRAQ5在含0.1%吐温-20 (v/v)的Odyssey缓冲液中),室温孵育1小时。用每孔115µL的缓冲液(1X PBS,含0.05%吐温-20 (v/v))洗涤5次,然后用每孔115µL的水洗涤3次。冲洗后,将平板倒置在薄的纸巾床上离心1分钟,转速高达1000 rpm,以去除多余的试剂,然后在室温下干燥,避免直接暴露在光线下。在Licor Odyssey系统上对底片成像,该系统测量700 nm和800 nm波长的综合强度。 为了抑制CYP1A2,在上述溶液中加入1 μL终浓度为40 μM的特异性药物底物(非那西丁:8 mM)。为了抑制CYP2B6,在上述溶液中加入1 μL的特异性药物底物(安非他酮:10 mM),终浓度为50 μM。为了抑制CYP2C8,在上述溶液中加入1 μL的特异性药物底物(紫杉醇:1 mM),终浓度为5 μM。为了抑制CYP2C9,在上述溶液中加入1 μL终浓度为200 μM的特异性药物底物(Tolbutamide: 40 mM)。为了抑制CYP2C19,在上述溶液中加入1 μL的特异性药物底物((s)-甲苯妥英:10 mM),终浓度为50 μM。为了抑制CYP2D6,在上述溶液中加入1 μL的特异性药物底物(右美沙芬:2 mM),终浓度为10 μM。为了抑制CYP3A4,在上述溶液中加入1 μL的特异性药物底物(咪达唑仑:1 mM),最终浓度为5 μM。37℃预热5 min,加入终浓度为1 mM的10 mM NADPH溶液20 μL, 37℃开始反应。在指定时间点加入400 μL冷淬溶液(甲醇含内标物[IS: 100 nM阿普唑仑,500 nM拉贝他洛尔,2 μM酮洛芬])停止反应(非那西丁:20 min;安非他酮:20分钟;紫杉醇:10分钟;甲苯磺丁胺:20分钟;(s)-甲苯妥英:20分钟;右美沙芬:20分钟;咪达唑仑:5分钟)。样品涡旋5分钟,4℃下3220 g离心40分钟。然后将100 μL上清液转移到加100 μL水的96孔板上(取决于LC-MS信号响应和峰形),进行LC-MS/MS分析。所有的实验都是重复进行的。[3] |
| 细胞实验 |
细胞增殖试验测定EZM0414在MM和DLBCL细胞系组中的ic50值。研究人员对EZM0414的肿瘤生长抑制(TGI)作用进行了评估。western blot检测H3K36me3水平,评价靶接合性。在7天的体外细胞实验中,评估了MM和DLBCL标准护理(SOC)药物联合抑制SETD2的潜力。[2]
以0.5 × 10-3 ~ 10 μmol/L的浓度依赖性处理对数生长期细胞系,最终DMSO浓度为0.2%体积/体积(v/v)。细胞以确定的(1.25 × 105个细胞/mL)电镀密度,一式三份,在96孔板上镀。实验板在37°C 5% CO2的湿化气氛中孵育。每隔3-4天,用钙素AM测定活细胞数,并在Acumen高含量成像仪上进行分析。细胞计数后,将细胞分裂回原电镀密度,更换生长培养基和化合物。用第14天的最终分裂调整活细胞数/mL计算抑制率。技术重复的抑制百分比平均值用于绘制浓度响应曲线,并计算每个时间点的绝对半最大抑制浓度(IC50)值。计算第4、7、11和14天的生长情况:1。计算第4 - 7天、第7 - 11天和第11-14天的分裂因子。分裂因子是第X天(4、7或11天)的活细胞/mL除以细胞被分裂回2的密度。对于第4天至第7天的细胞生长,将第7天的活细胞/mL密度乘以第4天的分裂因子。2. 对于第7天至第11天的细胞生长,将第11天的活细胞/mL密度乘以第4天和7个分裂因子。3. 对于第11天至第14天的细胞生长,将第14天的活细胞/mL密度乘以第4,7和11天的分裂因子。5. 在半对数图上绘制生长(Y轴为活细胞/mL,对数,X轴为天数)。[3] 以浓度依赖性浓度0.5 × 10-3 ~ 10 μmol/L的EZM0414处理对数相细胞系,最终DMSO浓度为0.2% v/v。将细胞以确定的镀密度在96孔板(1.25 × 105细胞/mL)中镀三次(用于第0 - 7天的疗程)或在6孔板(3.75 × 105细胞/mL)中镀(用于第7天的剩余时间的疗程)。实验板在37°C 5% CO2的湿化气氛中孵育。在第0、4、7天读取培养皿,在第4天补充化合物/培养基。第7天,将6孔板胰蛋白酶化,离心,重新悬浮在新鲜培养基中进行Vi-CELL计数。每个处理的细胞以原始密度重新镀于96孔板,一式三份,并以相同的化合物浓度重新处理。在第7天(播种第7天的另一个基线读数)、第11天和第14天读取培养皿,在第11天补充化合物/培养基。通过使用CellTiter-Glo®进行发光细胞活力测定,通过测量细胞腺苷-5 ' -三磷酸(ATP)来定量增殖,使用EnSpire多模微孔板阅读器检测发光。第14天的最终原始发光值用于计算抑制率。用技术重复的抑制百分比平均值绘制浓度响应曲线,并计算每个时间点的绝对IC50值。[3] 从培养箱中取出Caco-2板,用预温HBSS (10 mM HEPES, pH 7.4)洗涤2次,然后在37°C下孵育30分钟。对照化合物原液用DMSO稀释得到1 mM溶液,再用HBSS (10 mM HEPES, pH 7.4)稀释得到5 μM工作溶液。实验化合物的原液在DMSO中稀释得到1 mM溶液,然后用辅助信息稀释得到5 μM工作溶液。构像设计驱动发现EZM0414:一种选择性的、有效的SETD2抑制剂,用于临床研究SI-47 HBSS (10 mM HEPES, pH 7.4)。培养体系中DMSO的终浓度为0.5%。测定药物在根尖向基底外侧方向的转运速率。在Transwell插入物(顶端室)中加入75 μL的受试化合物和对照化合物的工作溶液,在接收板(基底外侧室)孔中填充235 μL的HBSS (10 mM HEPES, pH 7.4)。为测定药物在基底外侧至根尖方向的转运速率,将受试化合物和对照化合物的工作溶液分别取235 μL滴入受检板孔(基底外侧室),然后在Transwell插入物(根尖室)中填充75 μL HBSS (10 mM HEPES, pH 7.4)。在新的96孔板上,将10 μL工作溶液转移到40 μL HBSS (10 mM HEPES, pH 7.4)上,然后加入200 μL含有适当内标(IS)的冷乙腈或甲醇,制备Time 0样品。37℃孵育2小时。孵育结束时,将10 μL供体侧(A→B通量的根尖室,B→A的基底侧室)至40 μL HBSS (10 mM HEPES, pH 7.4)和50 μL受体侧(A→B通量的根尖室,B→A的根尖室)的样品转移到新的96孔板的孔中,然后加入4体积含有适当内标(IS)的冷乙腈或甲醇。样品涡旋5分钟,然后3220 g离心40分钟。在LC-MS/MS分析前,取上清液100µL与适当体积的超纯水混合。[3] |
| 动物实验 |
药代动力学[3]
研究设计:对动物进行单次静脉注射(IV)给药。同时,对禁食动物进行口服(PO)给药。在指定时间点,通过小鼠背侧跖静脉采集血液。将血液转移至含有K2-EDTA的采血管中。用于血浆分析的血液立即经离心处理以分离血浆,并储存在设定温度约为-80℃的冰箱中,直至分析。 样品制备:通过将待测物的储备液(1 mg/mL,溶于DMSO)用50%乙腈水溶液稀释,制备所需浓度范围的工作溶液。取10 μL工作溶液加入10 μL空白动物血浆中,配制浓度为0.5-1000 ng/mL的校准标准溶液,总体积为20 μL。将所得 20 µL 标准样品加入 200 μL 乙腈中进行蛋白质沉淀。所有样品均涡旋振荡 30 秒。在 4°C 和 4000 rpm(约 3740 xg)下离心 15 分钟后,取上清液并用水稀释。 分析方法:采用反相液相色谱-串联质谱法 (LC-MS/MS) 测定提取样品中的浓度。使用电喷雾电离 (ESI) 正离子模式下的多反应监测 (MRM) 监测分析物。峰面积由 Analyst® 软件积分,浓度通过峰面积比值(分析物峰面积/内标峰面积)与血浆校准标准品的理论浓度进行加权 (1/x2) 线性或二次回归确定。 药代动力学分析:采用非房室模型方法,使用线性/对数梯形法(静脉给药)或线性上升/对数下降梯形法(口服给药)(Phoenix WinNonlin 6.1,Certara,Princeton,NJ)分析小鼠的个体血浆浓度-时间数据。静脉给药后,计算清除率(CL)、稳态分布容积(Vss)、末端消除半衰期(t½)、曲线下面积(AUCINF)、平均共振时间(MRTINF)和末端分布容积(Vz)。口服给药后,计算最大观测浓度(Cmax)、达峰时间(tmax)、末端消除半衰期(t½)、曲线下面积(AUCINF)、平均共振时间(MRTINF)、表观总清除率(CL/F)、生物利用度(%F)和估计吸收分数(Fa)。 [3] 小鼠体内疗效分析 在研究开始前,动物被隔离7天。当平均肿瘤体积达到约119 mm³时,开始进行疗效研究的治疗。根据肿瘤体积,将小鼠随机分组,使各组的平均初始肿瘤体积相同。将溶于甲基纤维素/吐温-80水溶液(0.5% CMC/0.1% Tween-80 (Sigma-Aldrich))的EZM0414或仅含溶剂的EZM0414灌胃给NOD SCID小鼠(每剂量组n = 10)。[3] 对所有研究动物进行监测,不仅观察肿瘤生长情况,还观察其行为,例如活动能力、食物和水的消耗量、体重、眼/毛缠结情况以及任何其他异常反应。在整个疗效研究期间,每周两次使用游标卡尺测量肿瘤体积,并使用公式“肿瘤体积 = ax² + b²/2”估算肿瘤体积(mm³),其中“a”和“b”分别为肿瘤的长径和短径。肿瘤体积用于计算肿瘤生长抑制率(TGI,一种抗肿瘤疗效指标),计算公式为:TGI = (1-T/C) × 100%,其中“T”和“C”分别为肿瘤接种后某一天治疗组(T)和对照组(C)肿瘤的平均相对体积(肿瘤生长)。 |
| 药代性质 (ADME/PK) |
在小鼠中,EZM0414 和类似物 7 在 50 mg/kg 口服给药后均表现出良好的药代动力学特征,但 EZM0414 的暴露量(AUC)约为 7 的 2 倍,且口服生物利用度 (F) 也更高,这一趋势在大鼠中也观察到。表 3 列出了 EZM0414、7 和起始母体化合物 3 的药代动力学参数比较。早期系列化合物亲脂性的提高显著改善了整体药代动力学特征,在 CD-1 小鼠中,EZM0414 口服 50 mg/kg 后,其总体暴露量 (AUC0–∞) 提高了 17 倍。就药物相互作用作为CYP酶抑制剂的可能性而言,EZM0414仅对CYP同工酶2C8表现出较弱的抑制作用(4.8 μM),而对其他受试同工酶则未观察到抑制作用(同工酶1A2、2B6、2C9、2C19、2D6和3A4的IC50均>30 μM)。类似物7对同工酶2B6(3.2 μM)和2D6(10.8 μM)表现出中等程度的抑制作用,而对其余受试同工酶的IC50均>25 μM。对EZM0414的进一步表征表明其具有良好的安全性药理学特征。在由 47 个靶点组成的安全性小组和由 72 个激酶组成的多样性小组中对 EZM0414 进行体外测试,结果显示,除 D2(IC50 = 13.0 μM,拮抗剂)和 5-HT1B(IC50 = 3.2 μM,激动剂)外,所有靶点的 IC50 > 25 μM。[3]
|
| 参考文献 |
|
| 其他信息 |
SETD2抑制剂EZM0414是一种口服生物利用度高的选择性组蛋白甲基转移酶(HMT)SETD2(SET结构域蛋白2,组蛋白赖氨酸甲基转移酶)抑制剂,具有潜在的抗肿瘤活性。口服后,SETD2抑制剂EZM0414与SETD2结合并抑制其活性。这会阻止SETD2介导的多个关键生物学过程,包括组蛋白和非组蛋白的甲基化、转录调控、RNA剪接、DNA损伤修复以及B细胞的发育和成熟。EZM0414对SETD2的抑制可能抑制肿瘤细胞增殖。SETD2在肿瘤发生中发挥着多种重要作用。
引言:SETD2是目前已知唯一能够在体内催化H3K36三甲基化(H3K36me3)的组蛋白甲基转移酶(HMT)。 SETD2在多种生物学过程中发挥重要作用,包括B细胞的发育和成熟,由此推测,在这些过程中抑制SETD2可能具有抗肿瘤作用。B细胞的正常发育/成熟过程使其易受遗传脆弱性的影响,从而导致表观基因组失调和肿瘤发生,例如多发性骨髓瘤(MM)和弥漫性大B细胞淋巴瘤(DLBCL)。例如,15%-20%的MM携带高风险的t(4;14)染色体易位,导致多发性骨髓瘤SET结构域(MMSET)基因高表达。MMSET是一种组蛋白甲基转移酶(HMT),可催化H3K36me1和H3K36me2的形成,大量科学研究已证实MMSET过表达是t(4;14)骨髓瘤发病机制的关键因素。据我们所知,MMSET 一直是药物研发的靶点。然而,由于 t(4;14) 易位导致 SETD2 底物 H3K36me2 水平升高,抑制 SETD2 有望成为靶向 t(4;14) 多发性骨髓瘤 (MM) 患者中由 MMSET 过表达驱动的潜在致癌机制的有效手段。此外,迄今为止在白血病和弥漫性大B细胞淋巴瘤 (DLBCL) 中发现的 SETD2 功能缺失突变均为杂合突变,提示 SETD2 可能具有单倍体不足的抑癌作用。这一观察结果表明 SETD2 在白血病和淋巴瘤的生物学特性中发挥着关键作用,并提示 SETD2 抑制剂在这些或类似疾病中也可能具有治疗潜力。EZM0414 是一种首创的、高效、选择性强、口服生物利用度高的 SETD2 小分子酶抑制剂。我们利用 EZM0414 在多发性骨髓瘤 (MM) 和弥漫性大B细胞淋巴瘤 (DLBCL) 的临床前研究中探索了 SETD2 抑制剂的抗肿瘤作用,以验证其作为这些肿瘤类型治疗药物的潜力。[2] 在体外和体内临床前研究中,使用小分子抑制剂靶向 SETD2 可显著抑制 t(4;14) MM 以及非 t(4;14) MM 和 DLBCL 细胞系的生长。此外,体外实验观察到 EZM0414 与某些 MM 和 DLBCL 常用的标准治疗药物 (SOC) 具有协同作用,这支持将 SETD2 抑制剂与目前的 MM 和 DLBCL 疗法联合使用。这项工作为靶向治疗 B 细胞恶性肿瘤(如 MM,特别是 t(4;14) MM)以及 DLBCL 中的 SETD2 提供了理论依据,并为开展 1/1b 期临床研究以评估 EZM0414 在 R/R MM 和 DLBCL 患者中的安全性和活性奠定了基础。[2] |
| 分子式 |
C22H29FN4O2
|
|---|---|
| 分子量 |
400.49
|
| 精确质量 |
428.26
|
| 元素分析 |
C, 67.26; H, 7.76; F, 4.43; N, 13.07; O, 7.47
|
| CAS号 |
2411748-50-8
|
| 相关CAS号 |
EZM0414 TFA;2411759-92-5
|
| PubChem CID |
146395245
|
| 外观&性状 |
Off-white to light yellow solid powder
|
| LogP |
2.8
|
| tPSA |
68.4 Ų
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
29
|
| 分子复杂度/Complexity |
610
|
| 定义原子立体中心数目 |
2
|
| SMILES |
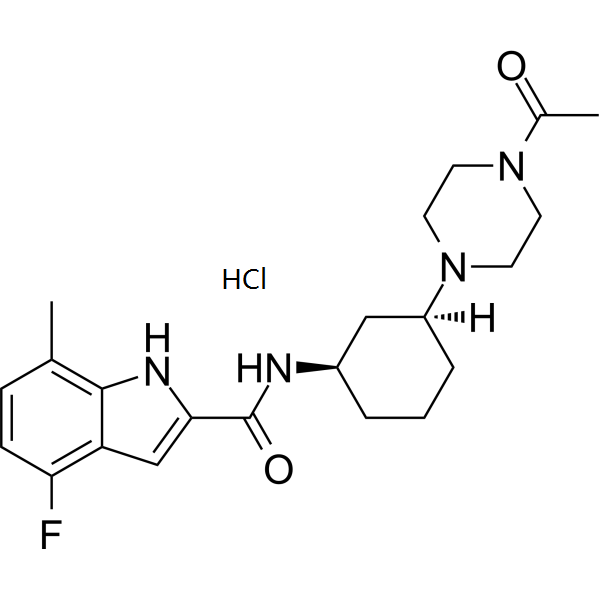
C1C=C(C2NC(=CC=2C=1F)C(N[C@@H]1CCC[C@H](N2CCN(CC2)C(=O)C)C1)=O)C
|
| InChi Key |
PGNLXEBQMQHFNK-SJORKVTESA-N
|
| InChi Code |
InChI=1S/C22H29FN4O2/c1-14-6-7-19(23)18-13-20(25-21(14)18)22(29)24-16-4-3-5-17(12-16)27-10-8-26(9-11-27)15(2)28/h6-7,13,16-17,25H,3-5,8-12H2,1-2H3,(H,24,29)/t16-,17+/m1/s1
|
| 化学名 |
N-[(1R,3S)-3-(4-acetylpiperazin-1-yl)cyclohexyl]-4-fluoro-7-methyl-1H-indole-2-carboxamide
|
| 别名 |
EZM0414 HCl; EZM-0414 HCl 2411748-50-8; KCY37T9RXU; EZM-0414; UNII-KCY37T9RXU; N-[(1R,3S)-3-(4-acetylpiperazin-1-yl)cyclohexyl]-4-fluoro-7-methyl-1H-indole-2-carboxamide; N-((1R,3S)-3-(4-Acetylpiperazin-1-yl)cyclohexyl)-4-fluoro-7-methyl-1H-indole-2-carboxamide; 1H-Indole-2-carboxamide, N-((1R,3S)-3-(4-acetyl-1-piperazinyl)cyclohexyl)-4-fluoro-7-methyl-;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮和光照。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4969 mL | 12.4847 mL | 24.9694 mL | |
| 5 mM | 0.4994 mL | 2.4969 mL | 4.9939 mL | |
| 10 mM | 0.2497 mL | 1.2485 mL | 2.4969 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 AOH-1996
AOH-1996
 hMAO-B-IN-3
hMAO-B-IN-3
 Flupyrimin
Flupyrimin
 阿曲库铵
阿曲库铵
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
