| 规格 | 价格 | ||
|---|---|---|---|
| 500mg | |||
| 1g | |||
| Other Sizes |

| 靶点 |
5-HT2B Receptor
|
|---|---|
| 体内研究 (In Vivo) |
给药 30 分钟后,甲基麦角新碱(0.2 mg/kg;静脉注射;单剂量)在家兔子宫、肝脏和肾脏中的平均浓度为 34 ng/g、80 ng/g 和 203 ng/g [3] 。甲基麦角新碱在家兔中的药代动力学 (PK) 参数 [3] = CO ng/mL K12/min K21/min Ke1/min 平均值 755.3 0.577 0.180 0.140 SD 215.8 0.098 0.036 0.069 T1/2α/min T1/2β/min Vdc L/kg Vdss L/kg Vdβ L/kg AUCtot (ng/mL)·min CLtot mL/min 平均值 0.91 26.34 0.29 1.13 1.44 6060.88 83.74 SD 0.35 9.48 0.09 0.30 0.55 2005.30 17.86
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Absorption is rapid after oral (60% bioavailability) and intramuscular (78% bioavailability) administration. Ergot alkaloids are mostly eliminated by hepatic metabolism and excretion, and the decrease in bioavailability following oral administration is probably a result of first-pass metabolism in the liver. 56.1 ± 0 L A delayed gastrointestinal absorption (Tmax about 3 hours) of methylergonovine tablets might be observed in postpartum women during continuous treatment with this oxytocic agent. Pharmacokinetic studies following an iv injection have shown that methylergonovine is rapidly distributed from plasma to peripheral tissues within 2-3 minutes or less. The bioavailability after oral administration was reported to be about 60% with no accumulation after repeated doses. During delivery, with intramuscular injection, bioavailability increased to 78%. Ergot alkaloids are mostly eliminated by hepatic metabolism and excretion, and the decrease in bioavailability following oral administration is probably a result of first-pass metabolism in the liver. Bioavailability studies conducted in fasting healthy female volunteers have shown that oral absorption of a 0.2 mg methylergonovine tablet was fairly rapid with a mean peak plasma concentration of 3243 +/- 1308 pg/mL observed at 1.12 +/- 0.82 hours. For a 0.2 mg intramuscular injection, a mean peak plasma concentration of 5918 +/- 1952 pg/mL was observed at 0.41 +/- 0.21 hours. The extent of absorption of the tablet, based upon methylergonovine plasma concentrations, was found to be equivalent to that of the i.m. solution given orally, and the extent of oral absorption of the i.m. solution was proportional to the dose following administration of 0.1, 0.2, and 0.4 mg. For more Absorption, Distribution and Excretion (Complete) data for METHYLERGONOVINE (15 total), please visit the HSDB record page. Metabolism / Metabolites Hepatic, with extensive first-pass metabolism. Ergot alkaloids are mostly eliminated by hepatic metabolism and excretion, and the decrease in bioavailability following oral administration is probably a result of first-pass metabolism in the liver. Hepatic, with extensive first-pass metabolism. Route of Elimination: Ergot alkaloids are mostly eliminated by hepatic metabolism and excretion, and the decrease in bioavailability following oral administration is probably a result of first-pass metabolism in the liver. Half Life: 3.39 hours Biological Half-Life 3.39 hours The plasma level decline was biphasic with a mean elimination half-life of 3.39 hours (range 1.5 to 12.7 hours). Plasma concentrations of methylergonovine appear to decline in a biphasic manner. Following IV administration of methylergonovine to adults with normal renal function, the half-life of the drug in the initial phase (t1/2 alpha?) reportedly ranges from about 1-5 minutes and the half-life in the terminal phase (t1/2 beta) ranges from about 0.5-2 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
Ergoline alkaloids have been shown to have the significant affinity towards the 5-HT1 and 5-HT2 serotonin receptors, D1 and D2 dopamine receptors, and alpha-adrenergic receptors. This can result in a number of different effects, including vasoconstriction, convulsions, and hallucinations. (A2914, A2915, A2916) Interactions Caffeine enhances action of ergot alkaloids in treatment of migraine... /Ergot alkaloids/ Peripheral and coronary vasoconstriction may be antagonized by nitrites or papaverine. /Ergot alkaloids/ Concomitant use of methylergonovine and inhibitors of cytochrome (CYP) 3A4 may result in vasospasm, cerebral ischemia, and/or ischemia of the extremities. Concomitant use of ergot alkaloids and human immunodeficiency virus (HIV) protease inhibitors, delavirdine, or nevirapine is contraindicated. Intravenous atropine sulphate following methylergonovine maleate administration may lead to severe hypertension and tachycardia. Non-Human Toxicity Values LD50 Mouse iv 85 mg/kg LD50 Rabbit iv 2600 mg/kg LD50 Mouse oral 187 mg/kg LD50 Rat iv 23 mg/kg LD50 Rat oral 93 mg/kg |
| 参考文献 |
[1]. Hajjo R, et al. Development, validation, and use of quantitative structure-activity relationship models of 5-hydroxytryptamine (2B) receptor ligands to identify novel receptor binders and putative valvulopathic compounds among common drugs. J Med Chem. 2010 Nov 11;53(21):7573-86.
[2]. Amant F, et al. Misoprostol compared with methylergometrine for the prevention of postpartum haemorrhage: a double-blind randomised trial. Br J Obstet Gynaecol. 1999 Oct;106(10):1066-70. [3]. Mäntylä R, et al. Plasma and tissue concentrations of methylergometrine (methylergonovine) in the rabbit. Acta Pharmacol Toxicol (Copenh). 1980 Apr;46(4):245-9. |
| 其他信息 |
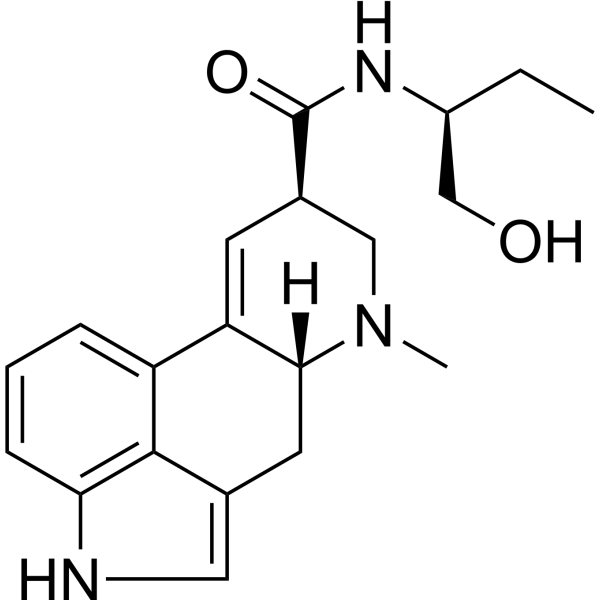
(6aR,9R)-N-[(2S)-1-hydroxybutan-2-yl]-7-methyl-6,6a,8,9-tetrahydro-4H-indolo[4,3-fg]quinoline-9-carboxamide is an ergoline alkaloid.
A homolog of ergonovine containing one more CH2 group. (Merck Index, 11th ed) Methylergonovine is an Ergot Derivative. Methylergonovine has been reported in Bos taurus with data available. Methylergonovine is only found in individuals that have used or taken this drug. It is a homolog of ergonovine containing one more CH2 group (Merck Index, 11th ed). Methylergonovine acts directly on the smooth muscle of the uterus and increases the tone, rate, and amplitude of rhythmic contractions through binding and the resultant antagonism of the dopamine D1 receptor. Thus, it induces a rapid and sustained tetanic uterotonic effect which shortens the third stage of labor and reduces blood loss. A homolog of ERGONOVINE containing one more CH2 group. (Merck Index, 11th ed) See also: Methylergonovine Maleate (has salt form); Methylergonovine tartrate (is active moiety of). Drug Indication For the prevention and control of excessive bleeding following vaginal childbirth Mechanism of Action Methylergometrine acts directly on the smooth muscle of the uterus and increases the tone, rate, and amplitude of rhythmic contractions through binding and the resultant antagonism of the dopamine D1 receptor. Thus, it induces a rapid and sustained tetanic uterotonic effect which shortens the third stage of labor and reduces blood loss. Methylergonovine acts directly on the smooth muscle of the uterus and increases the tone, rate, and amplitude of rhythmic contractions. Thus, it induces a rapid and sustained tetanic uterotonic effect which shortens the third stage of labor and reduces blood loss. Ergonovine maleate and methylergonovine maleate are pharmacologically similar. Both drugs directly stimulate contractions of uterine and vascular smooth muscle. Following administration of usual therapeutic doses of ergonovine or methylergonovine, intense contractions of the uterus are produced and are usually followed by periods of relaxation. Larger doses of the drugs, however, produce sustained, forceful contractions followed by only short or no periods of relaxation. The drugs increase the amplitude and frequency of uterine contractions and uterine tone which in turn impede uterine blood flow. Ergonovine and methylergonovine also increase contractions of the cervix. Ergonovine and methylergonovine produce vasoconstriction, mainly of capacitance vessels; increased central venous pressure, elevated blood pressure, and, rarely, peripheral ischemia and gangrene may result. Methylergonovine reportedly may interfere with prolactin secretion, but this effect has not been definitely established. Ergot alkaloids are antagonists of actions of 5-hydroxytryptamine and of certain metabolic actions of catecholamines. /Ergot alkaloids/ Therapeutic Uses Oxytocics For routine management after delivery of the placenta; postpartum atony and hemorrhage; subinvolution. Under full obstetric supervision, it may be given in the second stage of labor following delivery of the anterior shoulder. /Included in US product label/ Methylergonovine is a first-line agent for the treatment of postpartum hemorrhage; methylergonovine usually is given after oxytocin. Administration of parenteral ergot alkaloids during the third stage of labor decreases mean blood loss and the incidence of postpartum blood loss of 500 mL or more. /NOT included in US product label/ Ergonovine and methylergonovine should not be used for the induction or augmentation of labor. For more Therapeutic Uses (Complete) data for METHYLERGONOVINE (6 total), please visit the HSDB record page. Drug Warnings /Contraindications of methylergonovine therapy include:/ hypertension; toxemia; pregnancy; and hypersensitivity. Use of Methergine is contraindicated during pregnancy because of its uterotonic effects. This drug should not be administered iv routinely because of the possibility of inducing sudden hypertensive and cerebrovascular accidents. If iv administration is considered essential as a lifesaving measure, methylergonovine should be given slowly over a period of no less than 60 seconds with careful monitoring of blood pressure. Intra-arterial or periarterial injection should be strictly avoided. Caution should be exercised in the presence of sepsis, obliterative vascular disease, hepatic or renal involvement. Also use with caution during the second stage of labor. The necessity for manual removal of a retained placenta should occur only rarely with proper technique and adequate allowance of time for its spontaneous separation. For more Drug Warnings (Complete) data for METHYLERGONOVINE (13 total), please visit the HSDB record page. Pharmacodynamics Methylergometrine is a semisynthetic ergot alkaloid and a derivative of ergonovine and is used for the prevention and control of postpartum and post-abortion hemorrhage. In general, the effects of all the ergot alkaloids appear to results from their actions as partial agonists or antagonists at adrenergic, dopaminergic, and tryptaminergic receptors. The spectrum of effects depends on the agent, dosage, species, tissue, and experimental or physiological conditions. All of the alkaloids of ergot significantly increase the motor activity of the uterus. After small doses contractions are increased in force or frequency, or both, but are followed by a normal degree of relaxation. As the dose is increased, contractions become more forceful and prolonged, resting tonus is markedly increased, and sustained contracture can result. |
| 分子式 |
C20H25N3O2
|
|---|---|
| 分子量 |
339.43
|
| 精确质量 |
339.194
|
| CAS号 |
113-42-8
|
| PubChem CID |
8226
|
| 外观&性状 |
Prisms from methanol, acetone
Shiny crystals from benzene |
| 密度 |
1.3±0.1 g/cm3
|
| 沸点 |
638.4±55.0 °C at 760 mmHg
|
| 熔点 |
172ºC
|
| 闪点 |
339.9±31.5 °C
|
| 蒸汽压 |
0.0±2.0 mmHg at 25°C
|
| 折射率 |
1.665
|
| LogP |
1.67
|
| tPSA |
68.36
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
25
|
| 分子复杂度/Complexity |
549
|
| 定义原子立体中心数目 |
3
|
| SMILES |
CC[C@H](NC([C@H]1CN(C)[C@@]2(CC3=CNC4C3=C(C=CC=4)C2=C1)[H])=O)CO
|
| InChi Key |
UNBRKDKAWYKMIV-QWQRMKEZSA-N
|
| InChi Code |
InChI=1S/C20H25N3O2/c1-3-14(11-24)22-20(25)13-7-16-15-5-4-6-17-19(15)12(9-21-17)8-18(16)23(2)10-13/h4-7,9,13-14,18,21,24H,3,8,10-11H2,1-2H3,(H,22,25)/t13-,14+,18-/m1/s1
|
| 化学名 |
(6aR,9R)-N-[(2S)-1-hydroxybutan-2-yl]-7-methyl-6,6a,8,9-tetrahydro-4H-indolo[4,3-fg]quinoline-9-carboxamide
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.9461 mL | 14.7306 mL | 29.4612 mL | |
| 5 mM | 0.5892 mL | 2.9461 mL | 5.8922 mL | |
| 10 mM | 0.2946 mL | 1.4731 mL | 2.9461 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 463611831
463611831
