| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
Mite
|
|---|---|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Fenpyroximate was relatively well absorbed by rats after oral administration. Absorbed fenpyroximate was excreted predominantly via the biliary route, with lesser amounts in urine. The residual levels in organs and tissues after 168 hr were low. There was no evidence of bioaccumulation. Groups of four male Sprague-Dawley CD rats received single dermal applications of 14C-pyrazole-fenpyroximate suspended in water at doses of 0.1, 1.0, or 5.2 mg in 1 mL on a 10 sq cm area of skin for 0.5, 1, 2, 4, 10, or 24 hr and were sacrificed at the end of the exposure period, The concentration of radiolabel in blood was very low after all applications. Excretion in urine was slight but increased with duration of exposure; after 24 hr of exposure, 0.7-0.9% of the applied dose had been excreted. Radiolabel was found in feces after 10 and 24 hr of treatment, and fecal excretion was 1.4% at a dose of 1 mg, 0.5% at 10 mg, and 0.2% at 52 mg. These results suggest that fenpyroximate is barely absorbed from the skin and is excreted via the biliary-faecal and urinary routes. Groups of six male and six female Sprague-Dawley rats with bile duct cannulae were given a single oral dose of 2 mg/kg of (14)C-pyrazole- or (14)C-benzyl-fenpyroximate. Within 48 hr after treatment with pyrazole-labelled fenpyroximate, 47% (females) to 55% (males) of the radiolabel had been excreted in the bile, 5%, (males) to 10% (females) in urine, and 17% (females) to 28% (males) in feces. Total excretion 48 hr after treatment was about 88% for males and 73% for females. The Tmax, Cmax, and half-lives of the radiolabel in the blood of cannulated rats were similar to those of rats with no cannulae. Within 48 hr after oral administration of benzyl-labelled fenpyroximate, 47% (females) to 51% (males) of the radiolabel had been excreted in the bile, 6% (males) to 8% (females) in urine, and 28% (females) to 40% (males) in feces. Groups of six male and six female Sprague-Dawley (Crl;CD)rats were given single doses by gavage of 2 or 400 mg/kg bw of (3-(14)C)-pyrazole-(radiochemical purity, 96.4-99.9%) or (U-(14)C)-benzyl-fenpyroximate (radiochemical purity, 99.2-99.5%) suspended in 1% aqueous Tween 80. Blood was collected from the tail vein of five rats per group at various times up to 168 hrs after dosing. In rats given 2 mg/kg bw, the concentration of radiolabel in blood peaked within 1 hr after dosing and reached a plateau, which was sustained for about 18 hrs... In the group given 400 mg/kg bw, absorption was delayed, and radiolabel was not detectable in blood within the first 12 hrs after dosing. A nearly maximal level was achieved 12-24 hrs after dosing; the plateau was sustained for 80-100 hrs ... For more Absorption, Distribution and Excretion (Complete) data for Fenpyroximate (7 total), please visit the HSDB record page. Metabolism / Metabolites Fenpyroximate was extensively metabolized in rats; 23 metabolites were identified. No parent compound was found in the urine; metabolites found in the excreta represented 0-11% of the administered dose. Multiple pathways have been proposed for the metabolism of fenpyroximate, including oxidation, hydroxylation, demethylation, hydrolysis, and isomerization. Fenpyroximate is metabolized extensively by hydrolytic cleavage of the oxime ether bond, hydrolysis of the tert-butyl ester, oxidation of the tert-butyl, hydroxylation of the phenoxy ring and 3-methyl, isomerization, N-demethylation, and conjugation, producing a large number of metabolites. The major metabolites identified are (E)-4-[(1,3-dimethyl-5-phenoxypyrazol-4-yl) methyleneaminooxymethyl] benzoic acid, (Z)-4-[(1,3-dimethyl-phenoxypyrazol-4yl) methyleneaminooxymethyl] benzoic acid, (E)-4-{[1,3-dimethyl-5-(4-hydroxyphenoxy) pyrazol-4-yl]methyleneaminooxymethyl} benzoic acid, 1,3-dimethyl-5-phenoxypyrazole-4-carboxylic acid, 4-hydroxymethyl benzoic acid, terephthalic acid, 4-cyano-1-methyl-5-phenoxypyrazole-3-carboxylic acid, (E)-2-{4-[(1,3-dimethyl-5-phenoxypyrazol-4-yl) methyleneaminooxymethyl] benzoyloxy}-2-methylpropanoic acid, (E)-2-{4-[1,3-dimethyl-5-(4-hydroxyphenoxy) pyrazol-4-yl] methyleneaminooxymethyl} benzoyloxy]-2-methylpropionic acid, and (E)-2-[{4-[3-hydroxymethyl-1-methyl-5-phenoxypyrazol-4-yl] methyleneaminooxymethyl} benzoyloxy]-2-methylpropionic acid. Groups of six male and six female Sprague-Dawley rats with bile-duct cannulae were given a single oral dose of 2 mg/kg (14)C-pyrazole-labeled fenpyroximate. No parent fenpyroximate was found in bile, but metabolites (E)-4-[(1,3-dimethyl-5-phenoxypyrazol-4-yl)-methyleneaminooxy-methyl] benzoic acid, (Z)-4-[(1.3-dimethyl-5-phenoxy-pyrazol-4-yl)-methyleneaminooxy-methyl] benzoic acid, (E)-4-{[1,3-dimethyl-5-(4-hydroxyphenoxy) pyrazol-4-yl] methyleneaminooxymethyl} benzoic acid, (E)-2-{4-[(1,3-dimethyl-5-phenoxypyrazol- 4-yl) methyleneaminooxymethyl] benzoyloxy}-2-methylpropanoic acid, 1,3-dimethyl-5-(4-hydroxyphenoxy)pyrazole-4-carbaldehyde, 1,3-dimethyl-5-phenoxypyrazole-4-carboxylic acid, 3-methyl-5-phenoxypyrazole-4-carbaldehyde, 1,3-dimethyl-5-(4-hydroxyphenoxy)-pyrazole-4-carbonitrile, (E)-1,3-dimethyl-5-phenoxypyrazole-4-carbaldehydeoxime, 3-methyl-5-(4-hydroxyphenoxy)-pyrazole-4-carbaldehyde, and (E)-2-[4-[(1,3-dimethyl-5-phenoxy-pyrazol-4-yl)methyleneamniooxy-methyl]benzoyloxy]-2-methyl-propanoic acid, and conjugates of (E)-4-[(1,3-dimethyl-5-phenoxypyrazol-4-yl)-methyleneaminooxy-methyl] benzoic acid, (Z)-4-[(1.3-dimethyl-5-phenoxy-pyrazol-4-yl)-methyleneaminooxy-methyl] benzoic acid, (E)-4-{[1,3-dimethyl-5-(4-hydroxyphenoxy) pyrazol-4-yl] methyleneaminooxymethyl} benzoic acid, and 1,3-dimethyl-5-phenoxypyrazole-4-carboxylic acid were found. Total radiolabel represented less than 2% of the dose. The metabolic pathway proposed for fenpyroximate in rats is cleavage of the ester bond, hydroxylation at the phenoxypyrazole group, oxidation at the tert-butyl group, and conjugation with sulfate and glucuronide. Groups of four male Sprague-Dawley (SLC) rats were treated with a single oral dose of 1.5 mg/kg bw (14)C-pyrazole- or (14)C-benzoyl- labeled fenpyroximate (radioactive purity, > 99%), and urinary and fecal samples were collected for 0-72 hrs. Six urinary and 17 fecal metabolites were identified by thin-layer co-chromatography with authentic samples. ... The major urinary metabolites were 1,3-dimethyl-5-phenoxypyrazole-4-carboxylic acid (7.3% of the dose), 4-cyano-1-methyl-5-phenoxy-pyrazole-3-carboxylic acid (2.5%), and terephthalic acid (3.8%). The major fecal metabolites were (E)-4-[(1,3-dimethyl-5-phenoxypyrazol-4-yl)-methyleneaminooxy-methyl] benzoic acid (4.1-11.0% of the dose), (E)-4-{[1,3-dimethyl-5-(4-hydroxyphenoxy) pyrazol-4-yl] methyleneaminooxymethyl} benzoic acid (2.9-4.2%), and (E)-2-[4-[(1,3-dimethyl-5-phenoxy-pyrazol-4-yl)methyleneamniooxy-methyl]benzoyloxy]-2-methyl-propanoic acid (3.5-4.3%); 4-hydroxymethyl benzoic acid (7.5%) was found as a precursor of terephthalic acid and (E)-2-[4-[1,3-dimethyl-5-(4-hydroxyphenoxy) pyrazol-4-yl] methyleneaminooxymethyl} benzoyloxy]-2-methylpropionic acid (2.0-9.7%) and (E)-2-[{4-[3-hydroxymethyl-1-methyl-5-phenoxypyrazol-4-yl] methyleneaminooxymethyl} benzoyloxy]-2-methylpropionic acid (3.3-4.5%) as hydroxylated bodies of (E)-2-[4-[(1,3-dimethyl-5-phenoxy-pyrazol-4-yl)methyleneamniooxy-methyl]benzoyloxy]-2-methyl-propanoic acid. The concentrations of the urinary metabolites 1,3-dimethyl-5-(4-hydroxyphenoxy)-pyrazole-4-carbonitrile and 3-methyl-5-(4-hydroxyphenoxy)-pyrazole-4-carbaldehyde and the fecal metabolites tert-butyl(E)-4-[(1,3-dimethyl-5-(4-hydroxyphenoxy)pyrazol-4-yl)methyleneaminooxymethyl] benzoate, (E)-2-[4-[(1,3-dimethyl-5-phenoxy-pyrazol-4-yl)methyleneamniooxy-methyl]benzoyloxy]-2-methyl-propanoic acid, (E)-2-[4-[1,3-dimethyl-5-(4-hydroxyphenoxy) pyrazol-4-yl] methyleneaminooxymethyl} benzoyloxy]-2-methylpropionic acid, and (E)-2-[{4-[3-hydroxymethyl-1-methyl-5-phenoxypyrazol-4-yl] methyleneaminooxymethyl} benzoyloxy]-2-methylpropionic acid were increased by enzymatic hydrolysis of the excreta with beta-glucuronidase or sulfatase. In the present study, least 3 rats/sex/dose/time interval combination were treated with a single oral dose of (Pyrazole-(14)C) Fenpyroximate (NNI-850, purity: 99.6%), prior to collection of urine, feces, organic volatiles, and carbon dioxide. The preliminary study found no detectable carbon dioxide, and organic volatiles were either non-detectable or below quantifiable levels. ... Single oral doses were low (2 mg/kg) or high (400 mg/kg). Sacrifice time intervals after radiolabeled Fenpyroximate treatment were 12, 24, and 168 hr for low dose groups, and 12, 24, 96, 120, and 168 hr for high dose groups. Repeat dose groups received 2 mg/kg/day unlabeled Fenpyroximate for 14 days, followed by a single treatment with labeled Fenpyroximate at 2 mg/kg. This group was maintained for 168 hr before sacrifice. All 168-hr groups consisted of 5/sex, and these were used for excretion samples at intervals throughout that period. Metabolite identification was performed by comparisons of 2-dimensional TLC mobilities of excreta extracts with mobilities of a series of proposed metabolites in two sets of solvent systems (i.e. standard chromatograms by visualization under UV light were compared to autoradiograms of fecal or urinary extracts). ... A single 2 mg/kg dose found about 8% of fecal metabolites as parent compound, about 13% presumed to be the ester hydrolysis product, with other characterized metabolites accounting for about 5% or less of fecal radioactivity. Uncharacterized metabolites which remained at the origin of the 0-24 hr TLC plates of 2 mg/kg groups constituted 47-50% of fecal label, compared to 2-4% in 400 mg/kg groups, suggesting that most of the dose was absorbed and metabolized following low dose administration. ... The major identified urinary metabolite was evidently 1,3-dimethyl-5-phenoxypyrazol- 4-carboxylic acid. This compound, designated M-8, was substantially conjugated as a glucuronide. ... Biological Half-Life Groups of six male and six female Sprague-Dawley (Crl;CD)rats were given single doses by gavage of 2 or 400 mg/kg bw of (3-(14)C)-pyrazole-(radiochemical purity, 96.4-99.9%) or (U-(14)C)-benzyl-fenpyroximate (radiochemical purity, 99.2-99.5%) suspended in 1% aqueous Tween 80. Blood was collected from the tail vein of five rats per group at various times up to 168 hrs after dosing. In rats given 2 mg/kg bw ... slow elimination phase with a half-life of 6-9 hrs. In the group given 400 mg/kg bw ... elimination phase with a half-life of 35-49 hrs. Two studies used five rats/sex/group, dosed with 2 or 400 mg/kg (Pyrazole-(14)C) Fenpyroximate (NNI-850) or (Benzyl- (14)C) Fenpyroximate (NNI-850) by gavage in 1% aqueous Tween 80. Tail vein blood was collected at intervals for 7 days. Pyrazole-(14)C Study: Half-lives in blood were 8.9 hr for both M and F at 2 mg/kg. ... In contrast, 400 mg/kg led to half-lives in blood of 49 and 45 hr for M and F. ... Benzyl-(14)C Study: Half-lifes in blood were 6.1 hr and 7.9 hr for M and F, respectively, at 2 mg/kg. ... In contrast, 400 mg/kg led to half-lives in blood of 47 and 35 hr for M and F. |
| 毒性/毒理 (Toxicokinetics/TK) |
Non-Human Toxicity Values
LC50 Rat inhalation (female) 0.33 mg/L/4 hr /nose-only exposure/ LC50 Rat inhalation (male) 0.21 mg/L/4 hr /nose-only exposure/ LC50 Rat inhalation (female) 0.36 mg/L/4 hr /whole-body exposure/ LC50 Rat inhalation (male) 0.33 mg/L/4 hr /whole-body exposure/ For more Non-Human Toxicity Values (Complete) data for Fenpyroximate (9 total), please visit the HSDB record page. |
| 参考文献 |
|
| 其他信息 |
Fenpyroximate is a pyrazole acaricide and a tert-butyl ester. It has a role as a mitochondrial NADH:ubiquinone reductase inhibitor. It derives from a hydride of a 1H-pyrazole.
Fenpyroximate is under investigation in clinical trial NCT02533336 (The Effectiveness of Non-pyrethroid Insecticide-treated Durable Wall Liners as a Method for Malaria Control in Endemic Rural Tanzania). Mechanism of Action The high specificity of fenpyroximate as a miticide is not based primarily on differences in target site sensitivity since it inhibits the mitochondrial complex I from rat liver and from spider mites with less than a 10-fold difference in potency. The main mechanism of selectivity has been shown to depend on differential rates of metabolic detoxification, particularly through removal of the t-Bu group yielding the free carboxylic acid analog. This metabolite is inactive as a complex I inhibitor. This apparent hydrolysis is largely catalyzed by cytochrome PA50 through hydroxylation of the t-Bu group followed by intramolecular ester cleavage. Oxidative ester cleavage was rapid in the several mammals, fish, and insects tested but it did not occur in mites. Parkinson's disease (PD) brains show evidence of mitochondrial respiratory Complex I deficiency, oxidative stress, and neuronal death. Complex I-inhibiting neurotoxins, such as the pesticide rotenone, cause neuronal death and parkinsonism in animal models. We have previously shown that DJ-1 over-expression in astrocytes augments their capacity to protect neurons against rotenone, that DJ-1 knock-down impairs astrocyte-mediated neuroprotection against rotenone, and that each process involves astrocyte-released factors. To further investigate the mechanism behind these findings, we developed a high-throughput, plate-based bioassay that can be used to assess how genetic manipulations in astrocytes affect their ability to protect co-cultured neurons. We used this bioassay to show that DJ-1 deficiency-induced impairments in astrocyte-mediated neuroprotection occur solely in the presence of pesticides that inhibit Complex I (rotenone, pyridaben, fenazaquin, and fenpyroximate); not with agents that inhibit Complexes II-V, that primarily induce oxidative stress, or that inhibit the proteasome. This is a potentially PD-relevant finding because pesticide exposure is epidemiologically-linked with an increased risk for PD. Further investigations into our model suggested that astrocytic GSH and heme oxygenase-1 antioxidant systems are not central to the neuroprotective mechanism. ... In this study, ...the in vitro toxicity and mechanism of action of several putative complex I inhibitors that are commonly used as pesticides/ tebunfenpyrad. A similar order of potency was observed for reduction of ATP levels and competition for (3)H-dihydrorotenone (DHR) binding to complex I, with the exception of pyridaben (PYR). Neuroblastoma cells stably expressing the /rotenone/ (ROT)-insensitive NADH dehydrogenase of Saccharomyces cerevisiae (NDI1) were resistant to these pesticides, demonstrating the requirement of complex I inhibition for toxicity. ... PYR was a more potent inhibitor of mitochondrial respiration and caused more oxidative damage than ROT. The oxidative damage could be attenuated by NDI1 or by the antioxidants alpha-tocopherol and coenzyme Q(10). PYR was also highly toxic to midbrain organotypic slices. These data demonstrate that, in addition to ROT, several commercially used pesticides directly inhibit complex I, cause oxidative damage, and suggest that further study is warranted into environmental agents that inhibit complex I for their potential role in Parkinson's Disease. |
| 分子式 |
C24H27N3O4
|
|---|---|
| 分子量 |
421.49
|
| 精确质量 |
421.2
|
| CAS号 |
111812-58-9
|
| 相关CAS号 |
(E)-Fenpyroximate;134098-61-6
|
| PubChem CID |
9576412
|
| 外观&性状 |
White crystalline powder
|
| 密度 |
1.1±0.1 g/cm3
|
| 沸点 |
546.2±60.0 °C at 760 mmHg
|
| 熔点 |
99-102ºC
|
| 闪点 |
284.1±32.9 °C
|
| 蒸汽压 |
0.0±1.5 mmHg at 25°C
|
| 折射率 |
1.561
|
| LogP |
6.44
|
| tPSA |
74.94
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
31
|
| 分子复杂度/Complexity |
592
|
| 定义原子立体中心数目 |
0
|
| SMILES |
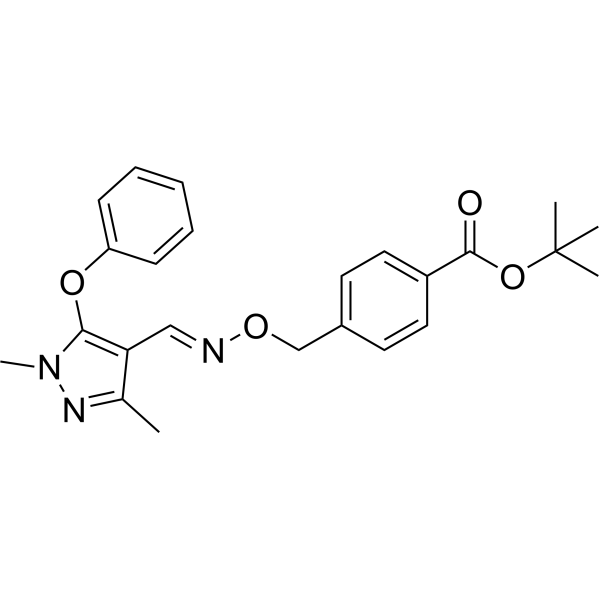
O(C1C=CC=CC=1)C1N(N=C(C=1C=NOCC1C=CC(=CC=1)C(=O)OC(C)(C)C)C)C
|
| InChi Key |
YYJNOYZRYGDPNH-MFKUBSTISA-N
|
| InChi Code |
InChI=1S/C24H27N3O4/c1-17-21(22(27(5)26-17)30-20-9-7-6-8-10-20)15-25-29-16-18-11-13-19(14-12-18)23(28)31-24(2,3)4/h6-15H,16H2,1-5H3/b25-15+
|
| 化学名 |
tert-butyl 4-[[(E)-(1,3-dimethyl-5-phenoxypyrazol-4-yl)methylideneamino]oxymethyl]benzoate
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3725 mL | 11.8627 mL | 23.7254 mL | |
| 5 mM | 0.4745 mL | 2.3725 mL | 4.7451 mL | |
| 10 mM | 0.2373 mL | 1.1863 mL | 2.3725 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Prostaglandin Bx
Prostaglandin Bx
 BE2647
BE2647
 SHO1122147
SHO1122147
 AK-4
AK-4
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
