| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 100mg |
|
||
| 500mg |
|
||
| 1g |
|
||
| Other Sizes |
|

| 靶点 |
DENV2 NS2B/NS3 protease (IC50 = 0.69 µM); ZIKV NS2B/NS3 protease (IC50 = 1.04 µM)
|
|---|---|
| 体外研究 (In Vitro) |
在这项工作中,研究人员阐明了基于苯并[d]噻唑的变构NS2B/NS3抑制剂的新的构效关系。他们开发了一系列新的y形抑制剂,其具有更大的疏水接触面,应该结合到以前未定位的变弹性NS2B/NS3结合口袋区域。通过支架跳跃,研究人员改变了苯并[d]噻唑核心,并确定苯并呋喃作为一种新的先导支架,将最初靶向zikv抑制剂的选择性转移到对DENV蛋白酶的更高活性。此外,研究人员能够通过随后的抑制剂截断将配体效率从0.27提高到0.41,并鉴定出N-(5,6-二羟基苯并[d]噻唑-2-基)-4-碘苯酰胺是一种新型亚微摩尔NS2B/NS3抑制剂。利用基于细胞的分析,研究人员可以证明纤维素中的抗病毒活性。总的来说,研究人员报道了一系列新的亚微摩尔变构DENV和ZIKV抑制剂,在细胞毒性和蛋白酶抑制选择性方面具有良好的疗效。[1]
|
| 酶活实验 |
荧光测定[1]
通过基于荧光底物或基于fret底物的测定来确定化合物对蛋白酶的抑制活性。抑制剂和底物作为原液在DMSO中制备。使用Tecan Infinite F2000 PRO读板仪在96孔白色平底微滴板上测量荧光。测量至少在三个独立的实验中进行。每孔的总容积为200µL,包括180µL缓冲液、5µL酶溶液、10µL DMSO或纯DMSO抑制剂作为对照,以及5µL相应底物溶液。抑制剂浓度为20µM时进行初始筛选。采用0.01µM ~ 100µM稀释系列测定IC50值。在25°C下,用相应的激发和发射波长每30 s测量一次荧光,持续10 min。通过将剩余酶活性拟合到四参数IC50方程中,用GraFit计算IC50值,其中Y [ΔF/min]为底物水解速率,Ymax为剂量-响应曲线的最大值,在抑制剂浓度[I] = 0µM时测量,Ymin为最小值,在高抑制剂浓度下获得,s为Hill系数。 |
| 细胞实验 |
复制分析和荧光素酶测定[1]
荧光素酶活性定量测定ZIKV和DENV2 RNA复制,如前所述。68,69简单地说,将Huh7细胞通过胰蛋白酶化制备单细胞悬液,用磷酸盐缓冲盐水洗涤一次。细胞以1 × 10~7个细胞/ mL的浓度重悬于含有2 mM ATP和5 mM谷胱甘肽的细胞液中。将10 μg体外转录的RNA与400 μL细胞悬浮液(4x10~6个细胞)混合,在975 μF、270 v的间隙宽度为0.4 cm的培养皿中,用Gene Pulser系统进行电穿孔转染。细胞在12孔板上的双孔中接种:时间点为4h和24h 500µL,时间点为48h, 72h和96h 1 mL。电穿孔后4 h,细胞用10µM化合物或等量DMSO作为对照,在添加15mm HEPES的培养基中处理。48 h后补充含化合物或dmso的培养基。在电穿孔后4、24、48、72和96 h,分别加入250µL荧光素酶裂解缓冲液(0.1% (v/v) TritonX-100, 25 mM甘氨酸,15 mM MgSO4, 15 mM K3PO4 pH 7.8, 4 mM EGTA, 10% (v/v)甘油和1 mM DTT)裂解细胞。为了检测豚鼠荧光素酶的活性,将100 μL裂解液与200 μL荧光素酶缓冲液(25 mM甘氨酸、15 mM MgSO4、15 mM K3PO4 pH 7.8和4 mM EGTA)混合,并添加14或28 nM coelanterazine (P.J.K)。为了检测萤火虫荧光素酶的活性,将100 μL裂解液与350 μL荧光素酶测定缓冲液混合,新鲜添加1 mM DTT和2 mM ATP,并将d-荧光素底物(200 μ M d-荧光素,p.j k)混合在25 mM甘氨酸中。 细胞活力测定[1] 为了确定化合物处理对细胞活力的影响,将Huh7细胞以每孔4 × 10~3个细胞的密度接种于白壁96孔板中,接种后1天,细胞分别用1.25、2.5、5、10、20和40µM化合物或同等体积的DMSO处理96小时。按照制造商的说明,使用CellTiter-Glo发光细胞活力测定法测量细胞活力。用平板光度计测定细胞活力,并将值归一化到未处理的细胞。 |
| 参考文献 | |
| 其他信息 |
In recent years, dengue virus (DENV) and Zika virus (ZIKV) – both mosquito-borne viruses of the Flaviviridae family – have become transcontinental health problems as climate change and globalization have led to the spread of their vectors from tropical to temperate regions. DENV and ZIKV are both positive-sense single-stranded RNA viruses whose genomes consist of three structural proteins (capsid protein, membrane precursor protein, and envelope protein) and seven non-structural proteins (NS proteins), all of which are initially expressed as a single precursor polyprotein. During viral maturation, the processing of the polyprotein is accomplished by the host protease and the viral NS2B/NS3 protease complex. Studies have shown that inhibitors of the NS2B/NS3 protease complex are effective antiviral drugs and can reduce viral pathogenicity. [1]
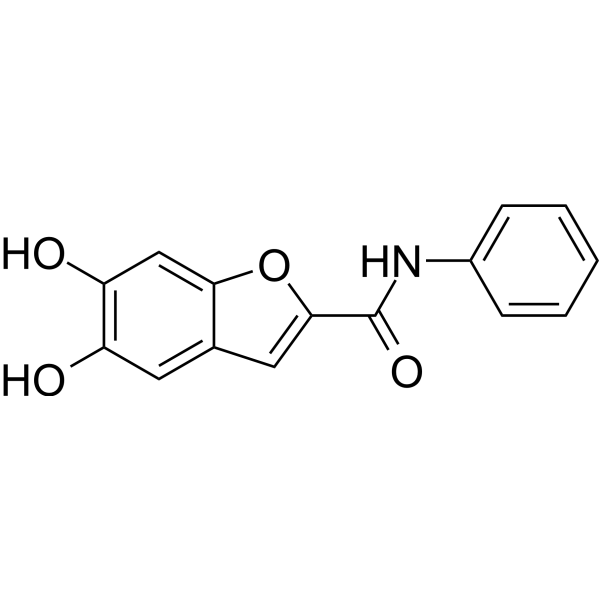
This study optimized allosteric inhibitors of DENV2 and ZIKV NS2B/NS3 proteases and discovered novel lead compounds with good inhibitory properties. Different series of inhibitors were synthesized by exchanging parts of lead compounds 1a and 1b, and their inhibitory effects on dengue virus type 2 (DENV2) and Zika virus (ZIKV) NS2B/NS3 proteases were investigated. Replacing the amino acid linker with (R/S)-alanine (8a, 8b), isoleucine (8c), tert-leucine (8d), phenylalanine (8e), tyrosine (12a), and aspartic acid (12b) did not significantly improve the inhibitory effect. However, compounds 8a–f and 12a inhibited the Zika virus protease in the low micromolar concentration range (IC50 = 2.13–6.48 µM). Among the newly designed Y-type inhibitor series (Table 2), the 2,2-diphenylacetic acid derivative 23b is the most promising compound, with an IC50 value for Zika virus (ZIKV) below the micromolar level (0.95 µM). The IC50 values of Y-type compounds 23b and 20b, as well as AA-type compounds 8c-f with hydrophobic side chains, are all in the low micromolar range, indicating that using Y-type inhibitors to simultaneously target two binding subsites is a promising strategy that could help further improve inhibitory efficacy. By truncating the inhibitor backbone (Table 3), we achieved, for the first time, low micromolar inhibition of dengue virus type 2 (DENV2) protease while improving ligand potency, providing a new starting point for further DENV2 drug development. The iodine-substituted inhibitor 25b is the most promising in this series, with an IC50 of 4.38 µM for DENV2 and a submicromolar IC50 (0.67 µM) for ZIKV. By replacing the benzo[d]thiazole core heterocycle, we demonstrated that other heteroaromatic ring systems besides benzo[d]thiazole can also serve as backbones. In this structure-activity relationship series, the benzofuran derivative 34e exhibited the best inhibitory activity (IC50(DENV2) = 0.69 µM, IC50(ZIKV) = 1.04 µM). Compared with benzo[d]thiazole compounds, 34e showed inhibitory activity against the NS2B/NS3 proteases of ZIKV and DENV2 on the same order of magnitude. Screening of the selected compounds against various serine and cysteine proteases demonstrated excellent off-target selectivity for our inhibitors. We employed a cell-based antiviral assay to investigate the potential of the most active inhibitors and their corresponding methoxy prodrugs to interfere with dengue virus type 2 (DENV2) and Zika virus (ZIKV) replication. Compounds 22b, 23b, and 25b significantly inhibited DENV2 replication, highlighting their antiviral potential; while compounds 25b and 34e reduced ZIKV replication. In conclusion, we identified two promising compounds that could serve as a suitable starting point for future drug development. First, N-(5,6-dihydroxybenzo[d]thiazolyl)-4-iodobenzamide (25b) and second, 5,6-dihydroxy-N-phenylbenzofuran-2-carboxamide (34e) contain only 20 and 21 heavy atoms, respectively, and have ligand efficiencies higher than 0.4, making them a good starting point for further development of NS2B/NS3 inhibitors. [1] |
| 分子式 |
C15H11NO4
|
|---|---|
| 分子量 |
269.25
|
| 精确质量 |
269.068
|
| CAS号 |
2910757-30-9
|
| PubChem CID |
163409047
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| LogP |
2.8
|
| tPSA |
82.7
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
4
|
| 可旋转键数目(RBC) |
2
|
| 重原子数目 |
20
|
| 分子复杂度/Complexity |
356
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
IWQSFILCQBTRKR-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C15H11NO4/c17-11-6-9-7-14(20-13(9)8-12(11)18)15(19)16-10-4-2-1-3-5-10/h1-8,17-18H,(H,16,19)
|
| 化学名 |
5,6-dihydroxy-N-phenyl-1-benzofuran-2-carboxamide
|
| 别名 |
NS2B/NS3-IN-4; CHEMBL4865575; 2910757-30-9;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.7140 mL | 18.5701 mL | 37.1402 mL | |
| 5 mM | 0.7428 mL | 3.7140 mL | 7.4280 mL | |
| 10 mM | 0.3714 mL | 1.8570 mL | 3.7140 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 SARS-CoV-2 main protease
SARS-CoV-2 main protease
 AH-D peptide TFA
AH-D peptide TFA
 AG-1859
AG-1859
 NS2B/NS3-IN-9
NS2B/NS3-IN-9
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
