| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| Other Sizes |

| 靶点 |
P2Y13 receptor (pIC50= 5.97)
|
|---|---|
| 体外研究 (In Vitro) |
2-氯-5-硝基相似物(MRS 2211)和4-氯-3-硝基类似物(MRS 2603)抑制ADP(100 nM)诱导的肌醇三磷酸(IP3)形成,pIC50值分别为5.97和6.18,比PPADS强45倍和74倍。MRS 2211的拮抗作用具有竞争性,pA2值为6.3。MRS2211和MRS2603抑制了人P2Y1受体介导的1321N1星形细胞瘤细胞对30 nM 2-甲硫基-ADP的磷脂酶C(PLC)反应,IC50值分别为>10和0.245μM。作为1321N1星形细胞瘤细胞中人P2Y12受体介导的PLC反应的拮抗剂,这两种类似物均无活性(IC50>10μM)。因此,与P2Y1和P2Y12受体相比,MRS2211作为P2Y13受体的拮抗剂显示出>20倍的选择性,而MRS2603拮抗了P2Y1和P213受体[1]。
|
| 酶活实验 |
三磷酸肌醇(IP3)测定[1]
如上所述测量三磷酸肌醇的形成。简而言之,在实验前两天,将hP2Y13-AG32细胞(200000个细胞/孔)接种在直径35mm的完整培养基中的细胞培养皿上。实验前一天,用添加了5%FBS(v/v)、抗生素、G418和[myo-D-2-3H]-肌醇(5μCi/ml)的DMEM更换培养基。18小时后,吸出该标记培养基,将细胞在克雷布斯-林格-HEPES(KRH)缓冲液(124 mM NaCl、5 mM KCl、1.25 mM MgSO4、1.45 mM CaCl2、1.25 mM KH2PO4、25 mM HEPES、pH 7.4和8 mM D-葡萄糖)中再孵育2小时。然后,在37°C下用拮抗剂溶液或缓冲液单独预处理细胞10分钟,然后加入ADP 30秒。抽吸培养基并加入1ml 3%冰冷的高氯酸溶液后终止反应。然后,如前所述,在Dowex柱上分离三磷酸肌醇[18]。如报告所述,测量了表达人P2Y1受体的1321N1细胞中磷脂酶C(PLC)对2-甲硫基ADP反应的抑制,并对人P2Y12受体介导的反应使用了类似的方法。 |
| 细胞实验 |
细胞培养[1]
使用先前构建的表达Gα16蛋白并稳定转染hP2Y13受体(hP2Y13-1321N1-Gα16)的人星形细胞瘤1321N1细胞。细胞在添加了10%胎牛血清(FBS;v/v)、100单位/ml青霉素、100μg/ml链霉素、400μg/ml G418和500μg/ml zeocin的Dulbecco's Modified Eagle's Medium(DMEM)中培养。细胞在37°C下在9厘米培养皿中95%空气和5%二氧化碳的加湿气氛中生长。 |
| 参考文献 | |
| 其他信息 |
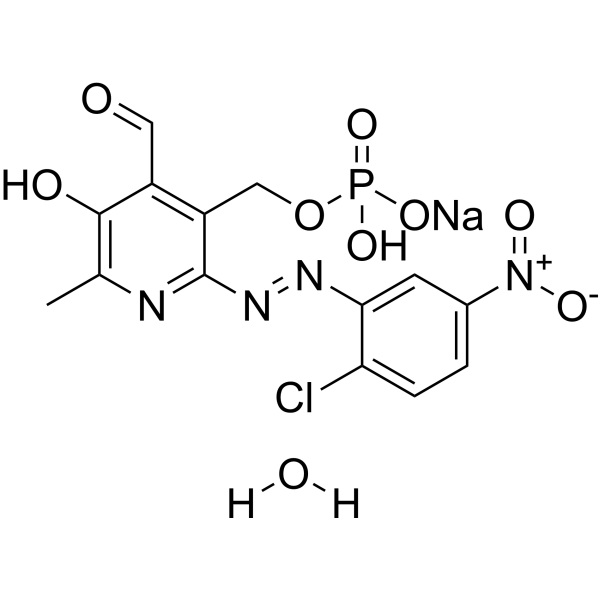
Researchers have synthesized a series of derivatives of the known P2 receptor antagonist PPADS (pyridoxal-5'-phosphate-6-azo-phenyl-2,4-disulfonate) and examined their ability to inhibit functional activity of the recombinant human P2Y13 nucleotide receptor expressed in 1321N1 human astrocytoma cells co-expressing G(alpha)16 protein (AG32). Analogues of PPADS modified through substitution of the phenylazo ring, including halo and nitro substitution, and 5'-alkyl phosphonate analogues were synthesized and tested. A 6-benzyl-5'-methyl phosphonate analogue was prepared to examine the effect of stable replacement of the azo linkage. The highest antagonistic potency was observed for 6-(3-nitrophenylazo) derivatives of pyridoxal-5'-phosphate. The 2-chloro-5-nitro analogue (MRS 2211) and 4-chloro-3-nitro analogue (MRS 2603) inhibited ADP (100 nM)-induced inositol trisphosphate (IP3) formation with pIC50 values of 5.97 and 6.18, respectively, being 45- and 74-fold more potent than PPADS. The antagonism of MRS 2211 was competitive with a pA2 value of 6.3. MRS2211 and MRS2603 inhibited phospholipase C (PLC) responses to 30 nM 2-methylthio-ADP in human P2Y1 receptor-mediated 1321N1 astrocytoma cells with IC50 values of >10 and 0.245 microM, respectively. Both analogues were inactive (IC50>10 microM) as antagonists of human P2Y12 receptor-mediated PLC responses in 1321N1 astrocytoma cells. Thus, MRS2211 displayed >20-fold selectivity as antagonist of the P2Y13 receptor in comparison to P2Y1 and P2Y12 receptors, while MRS2603 antagonized both P2Y1 and P2Y13 receptors.
The most potent antagonists among this series were two compounds containing both chloro and the m-nitro group in the phenylazo ring: 2-chloro-5-nitro (12, MRS 2211) and 4-chloro-3-nitro (13, MRS 2603). The inhibition by these compounds of the ADP (100 nM)-induced inositol trisphosphate synthesis was already evident at high nanomolar concentrations. Compounds 12 and 13 were 45- and 74-fold, respectively, more potent at the P2Y13 receptor than the parental compound PPADS. The interaction of MRS 2211 with the P2Y13 receptor had a competitive character with a pA2 value of 6.3, calculated from the Schild plot. [1] |
| 分子式 |
C14H13CLN4NAO9P
|
|---|---|
| 分子量 |
470.69
|
| 精确质量 |
473.972
|
| CAS号 |
1197030-56-0
|
| PubChem CID |
136068412
|
| 外观&性状 |
Solid powder
|
| tPSA |
193
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
11
|
| 可旋转键数目(RBC) |
5
|
| 重原子数目 |
30
|
| 分子复杂度/Complexity |
632
|
| 定义原子立体中心数目 |
0
|
| InChi Key |
VLNWYJFCKRYEJA-UHFFFAOYSA-L
|
| InChi Code |
InChI=1S/C14H12ClN4O8P.2Na/c1-7-13(21)9(5-20)10(6-27-28(24,25)26)14(16-7)18-17-12-4-8(19(22)23)2-3-11(12)15;;/h2-5,21H,6H2,1H3,(H2,24,25,26);;/q;2*+1/p-2
|
| 化学名 |
disodium;[2-[(2-chloro-5-nitrophenyl)diazenyl]-4-formyl-5-hydroxy-6-methylpyridin-3-yl]methyl phosphate
|
| 别名 |
MRS 2211; 2-[(2-CHLORO-5-NITROPHENYL)AZO]-5-HYDROXY-6-METHYL-3-[(PHOSPHONOOXY)METHYL]-4-PYRIDINECARBOXALDEHYDE DISODIUM SALT; 1197030-56-0; disodium;[2-[(2-chloro-5-nitrophenyl)diazenyl]-4-formyl-5-hydroxy-6-methylpyridin-3-yl]methyl phosphate; SCHEMBL3361090; CHEMBL1909455;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中,避免吸湿/受潮。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O : 32.95 mg/mL (70.00 mM; with sonication and heat)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1245 mL | 10.6227 mL | 21.2454 mL | |
| 5 mM | 0.4249 mL | 2.1245 mL | 4.2491 mL | |
| 10 mM | 0.2125 mL | 1.0623 mL | 2.1245 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
