| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 10 mM * 1 mL in DMSO |
|
||
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
BTK (IC50 = 37.9 nM)
Human Bruton’s Tyrosine Kinase (BTK) (IC50 = 1.6 nM, determined by kinase activity assay; covalent binding to Cys481) [1] - Murine BTK (IC50 = 3.2 nM, determined by kinase activity assay) [1] - Other TEC family kinases (ITK: IC50 = 45 nM; TXK: IC50 = 120 nM) [1] - Off-target kinases (EGFR, ERBB2, JAK3: IC50 > 1000 nM) [1] |
|---|---|
| 体外研究 (In Vitro) |
Evobrutinib 能够阻断 BTK 活性并阻止 BCR 信号通路被激活。它通过 GSH 结合、O-脱烷基化、羟基化、水解和葡萄糖醛酸化进行分解[2]。
Evobrutinib是一种新型、高选择性、不可逆的BTK抑制剂,可有效阻断Fc和BCR受体介导的信号传导。Evobrutinib具有阻断BTK活性和阻止BCR信号通路被激活的能力。它通过谷胱甘肽偶联、o -脱烷基、羟基化、水解和葡萄糖醛酸化分解[2]。
Evobrutinib是一种新型、高选择性、不可逆的BTK抑制剂,可有效抑制BCR-和Fc受体介导的信号传导,从而抑制人类B细胞和先天免疫细胞(如单核细胞和嗜碱性细胞)的后续激活和功能。[3] 强效选择性BTK抑制:依鲁替尼布(Evobrutinib; M2951)共价结合BTK的Cys481位点,抑制人BTK的IC50 = 1.6 nM,鼠BTK的IC50 = 3.2 nM;对ITK/TXK的选择性>60倍,对脱靶激酶的选择性>600倍[1] - 阻断B细胞受体(BCR)介导的信号通路:10 nM 依鲁替尼布(Evobrutinib; M2951)使人B细胞中BTK磷酸化(Tyr223)降低约90%,AKT磷酸化(Ser473)降低约85%;抑制抗IgM诱导的B细胞增殖,IC50 = 8.5 nM[1, 3] - 抑制免疫细胞活化:20 nM 依鲁替尼布(Evobrutinib; M2951)使人单核细胞中LPS诱导的TNF-α和IL-6分泌分别减少约70%和65%;在混合淋巴细胞反应中减少CD4+ T细胞增殖约50%[3] - 无显著细胞毒性:浓度高达100 μM时,人B细胞、单核细胞和T细胞的存活率>90%[1, 3] |
| 体内研究 (In Vivo) |
在这项研究中,研究人员在RA和SLE的临床前模型中评估了Evobrutinib/依沃鲁替尼,并描述了BTK占用与疾病活性抑制之间的关系。在类风湿性关节炎和SLE小鼠模型中,口服Evobrutinib显示出强大的疗效,证明了疾病严重程度和组织学损伤的降低。在SLE模型中,Evobrutinib抑制B细胞活化,降低自身抗体产生和浆细胞数量,并使B细胞和T细胞亚群正常化。在RA模型中,尽管未能降低自身抗体,但仍能达到疗效。药代动力学/药效学模型显示,在RA和SLE小鼠模型中,血细胞中BTK的平均占用率为80%与近乎完全的疾病抑制有关。此外,Evobrutinib在被动皮肤过敏反应模型中抑制肥大细胞活化。因此,Evobrutinib通过同时作用于B细胞和先天免疫细胞来达到疗效。综上所述,我们的数据表明,Evobrutinib是一种很有前途的分子,可用于B细胞驱动的自身免疫性疾病的慢性治疗。
小鼠实验性自身免疫性脑脊髓炎(EAE)模型的药效:口服依鲁替尼布(Evobrutinib; M2951)(10、30 mg/kg/天),剂量依赖性降低EAE临床评分约40%和65%;减少脊髓炎症和脱髓鞘程度55-70%[3] - 抑制大鼠B细胞活化:口服30 mg/kg 依鲁替尼布(Evobrutinib; M2951),使抗IgM诱导的脾B细胞增殖减少约75%,血清IgM水平降低约60%[1] - 胶原诱导关节炎(CIA)模型的药效:小鼠口服30 mg/kg/天,持续21天,足肿胀减轻约60%,关节组织病理学评分降低约55%[3] - 降低促炎细胞因子水平:30 mg/kg剂量使EAE小鼠血清TNF-α、IL-6和IFN-γ水平降低50-60%[3] |
| 酶活实验 |
激酶检测[3]
利用纯化的全长重组BTK测定依沃鲁替尼对BTK的效价。BTK蛋白用75 μM ATP和1 μM KinKDR肽FITC-AHA-EEPLYWSFPAKKK-NH2在缓冲液中稀释至终浓度0.05 ng/μl。不同浓度的依伏鲁替尼也包括在内。在25°C下反应90分钟,加入含有0.5 M EDTA的停止溶液停止反应。然后在Caliper LabChip 3000上读取板,并将数据加载到Genedata Screener中生成IC50曲线。为了比较evobrutinib或ibrutinib对野生型(WT) BTK与C481S BTK的抑制作用,我们使用了覆盖激酶结构域的重组蛋白(BTK WT 328-659或BTK C481S 328-659)。跳跃稀释实验,将含有100倍标准生化试验浓度BTK (0.63 nM)的20 μl实验缓冲液加入到200 nl依沃鲁替尼或RN486中,最终浓度为10倍IC50或阴性对照(DMSO)。室温孵育90 min后,将1 μl的混合溶液稀释到含有底物肽(序列FITC-AHA-EEPLYWSFPAKKK-NH2, 1 μM)和ATP (75 μM)的99 μl实验缓冲液中。将微孔板放置在Caliper Life Sciences LabChip 3000中,重复取样112分钟。在KinaseProfiler筛选板(EMD Millipore, Billerica, MA)中确定evobrutinib和ibrutinib的激酶选择性,测试化合物在1 μM下对267种激酶的抑制活性。[3] Ramos细胞BTK磷酸化/BTK phosphorylation in Ramos cells[3] 在Ramos B细胞中测定依维鲁替尼对BCR激活后BTK磷酸化的影响。Ramos Burkitt淋巴瘤细胞系从美国类型培养收集中获得,并在含有青霉素/链霉素,2mm l-谷氨酰胺和10%胎牛血清的RPMI 1640培养基中维持。将Ramos细胞接种于96孔组织培养板中,每孔8 × 106个细胞。用溶解在DMSO中的BTK抑制剂evobrutinib在37℃下预处理细胞30分钟。复合处理后,用浓度为5 μg/ml的抗igm抗体F(ab’)2 ab (SouthernBiotech, Birmingham, AL)刺激细胞激活BCR。细胞与抗igm在37℃下孵育5 min。处理后,收集细胞,500 × g离心5 min,抽吸培养基,加入150 μl含有Thermo Scientific Halt蛋白酶/磷酸酶抑制剂混合物的冰凉Thermo Scientific Pierce M-PER裂解缓冲液。将细胞重悬于裂解缓冲液中,裂解物冷冻至- 80°C,用于后续测量BTK磷酸化。根据制造商的说明,使用自动Wes仪器进行蛋白印迹分析BTK磷酸化。Western blot检测中,使用6 μl裂解液,用1:3000稀释的一抗BTK p-Y551 或1:50稀释的抗BTK p-Y223检测磷酸化的BTK。 BTK激酶活性测定:重组人/鼠BTK与ATP(含[γ-32P]ATP)、肽底物(BTK特异性)及系列稀释的依鲁替尼布(Evobrutinib; M2951)(0.001-100 nM)在激酶缓冲液中孵育。30°C孵育60分钟后,用酸性溶液终止反应。磷酸化肽段捕获于磷酸纤维素滤膜,定量放射性强度,从浓度-效应曲线计算IC50值[1] - 激酶选择性测定:45种激酶(包括TEC家族、EGFR、JAK3)在最适条件下,分别与各自底物、ATP及依鲁替尼布(Evobrutinib; M2951)(1 μM)孵育。放射分析法检测激酶活性,评估脱靶抑制作用[1] - 共价结合实验:重组BTK与依鲁替尼布(Evobrutinib; M2951)(0.1-10 μM)37°C孵育2小时,SDS-PAGE电泳后,用Cys481结合特异性抗体进行western blot,验证共价相互作用[1] |
| 细胞实验 |
外周血和全血中B细胞的活化[3]
Evobrutinib阻断BCR信号的能力是用全血B细胞或纯化的PBMCs来测定的。用柠檬酸盐作为抗凝血剂采集人血。对于PBMC实验,通过Ficoll梯度分离PBMC,每孔2.5 × 105个细胞接种到96孔板中。全血测定时,每孔90 μl的血直接转移到96孔板上。细胞在37℃用依沃鲁替尼稀释液预处理60 min,然后用山羊抗人IgM F(ab’)2 (Dianova)活化,加入终浓度为20 μg/ml, 37℃孵育过夜。活化后,细胞用抗cd69 - allophycocyanin和抗cd19 - percp - cy5.5染色45分钟,然后用FACS裂解液裂解,在PBS中重悬,然后进行FACS分析。在FACSCanto II仪器上进行FACS分析。首先对细胞进行CD19门控,并测定CD19阳性细胞的百分比。 B细胞增殖、细胞因子释放和质母细胞分化的研究[3] 使用B细胞纯化试剂盒II,按照制造商的说明,通过阴性选择从健康志愿者的外周血中分离CD19+ B细胞。纯化后的B细胞用evobrutinib孵育1 h,用10 μg/ml山羊F(ab’)2抗igm和10 ng/ml重组人IL-4刺激4 d,最后18 h加入[3H]胸腺嘧啶微孔孵育1 h。使用多板β计数器测量增殖。在细胞因子释放实验中,将健康志愿者外周血中分离的CD19+ B细胞与依维鲁替尼孵育1小时,然后用10 μg/ml兔抗人IgA + IgG + IgM (h +L)、3 μg/ml CpG寡脱氧核苷酸2006和8000 IU/ml重组人IFN-α刺激48小时,用细胞计数头阵列试剂盒检测上清中的细胞因子。为了产生Ig,分离的B细胞分别用20 U/ml IL-2、100 ng/ml IL-10、10 μg/ml灭活的金黄色葡萄球菌Cowan和不同浓度的依维鲁替尼刺激。培养10 d后,ELISA检测上清液中IgG和IgM水平。 fc - γ - r信号的抑制作用[3] U937 NF -κB-Luc报告细胞在二氧化碳调节的组织培养箱中37°C保持贴壁培养。实验当天,收集细胞,计数,并在96孔组织板中镀。Evobrutinib以5 nM ~ 10 μM的浓度加入。细胞与evobrutinib在37°C组织培养箱中孵育30分钟。然后将细胞转移到涂有抗cd64的新鲜板上并刺激4小时。使用EnVision板阅读器测量细胞裂解物中的荧光素酶活性。 嗜碱性粒细胞抑制试验[3] Evobrutinib阻断Fc受体信号的能力通过全血嗜碱性粒细胞测定。用柠檬酸盐作为抗凝剂收集人血,并转移到96孔板上。37℃用依维鲁替尼稀释液预处理30 min后,抗ige活化,加入终浓度2 μg/ml, 37℃孵育5 min。活化后,细胞用抗cd63 - fitc染色15 min,然后加入PBS-EDTA (20 mM),再加入固定/裂解缓冲液。在FACS分析之前,将细胞固定在甲醛中。使用FACSCanto II仪器对CD123+HLA-DR -细胞进行首次门联后,测定CD63表达的平均荧光强度(MFI)。[3] BioMAP分析[3] Evobrutinib和ibrutinib的生物选择性在体外使用原代人细胞和BioSeek的BioMAP分析进行评估。根据先前发表的详细方法,在12种不同的原代细胞共培养系统中,使用370 nM-10 μM的浓度范围对化合物的活性进行了评估。 B细胞增殖与信号通路实验:分离人外周血B细胞,接种于96孔板。用依鲁替尼布(Evobrutinib; M2951)(0.01-100 nM)预处理1小时,再用抗IgM(10 μg/mL)刺激72小时。MTT法检测细胞增殖;western blot检测BTK/AKT磷酸化水平[1, 3] - 单核细胞细胞因子分泌实验:人单核细胞用依鲁替尼布(Evobrutinib; M2951)(0.1-100 nM)预处理2小时,再用LPS(1 μg/mL)刺激24小时。收集培养上清液,ELISA法定量TNF-α/IL-6水平[3] - 混合淋巴细胞反应实验:人CD4+ T细胞与同种异体树突状细胞共培养,加入依鲁替尼布(Evobrutinib; M2951)(0.1-50 nM),培养5天。[3H]-胸腺嘧啶掺入法检测T细胞增殖[3] |
| 动物实验 |
DBA/1J雌性小鼠
12 mg/kg o.g. 小鼠全血中体外B细胞刺激[3] Evobrutinib通过灌胃法给予C57BL/6雌性小鼠(每组5只),剂量和时间点如所示。随后采集肝素化全血,并将其分成两份。一份与抗IgD抗体孵育作为刺激,另一份与PBS孵育作为基础对照。采用流式细胞术检测B细胞表面CD69的平均荧光强度(MFI)。计算刺激后MFI与基础MFI的差异,并以ΔMFI表示。抑制率按以下公式计算:抑制率 = (1− [Δ MFIevobrutinib/Δ MFIvehicle]) × 100。[3] PK/PD 模型和全血占有率测定[3] 为了构建evobrutinib的 PK/PD 关系,对 11-12 周龄的 DBA/1J 雌性小鼠进行给药。随时间推移测量血液中 BTK 的占有率和血浆浓度。Evobrutinib 配制成 20% kleptose 和 50 mM 柠檬酸盐(pH 3)的赋形剂溶液,并通过灌胃法给药。给药后不同时间点处死小鼠,并通过下腔静脉采集血液至肝素化试管中。为了测定 BTK 占有率,我们采用了一种先前描述的方法,该方法使用生物素标记的 BTK 结合探针和链霉亲和素捕获 ELISA。血浆样本通过液相色谱-串联质谱法分析,以测定 evobrutinib 的浓度。[3] 小鼠被动皮肤过敏反应[3] C57BL/6 小鼠背部皮内注射 250 ng 抗 DNP IgE 或抗 OVA IgE 进行致敏。24 小时后,所有小鼠均静脉注射 0.5 mg DNP-人血清白蛋白(含 0.5% 伊文思蓝)。激发后 30 分钟处死小鼠,取背部皮肤,并在 55°C 下用甲酰胺提取 24 小时以去除伊文思蓝。在 620 nm 波长处测定吸光度 (OD),并与已知浓度伊文思蓝的标准曲线进行比较。结果以每毫克组织中染料的纳克数表示。[3] 小鼠胶原诱导性关节炎[3] 所有小鼠胶原诱导性关节炎 (CIA) 研究均在 Bolder BioPATH 进行。DBA/1OlaHsd 小鼠(每组 12-15 只)用异氟烷麻醉,剃除尾根部毛发,并在第 0 天和第 21 天分别于尾根部皮内注射 150 μl 含有牛 II 型胶原蛋白 (2 mg/ml) 的 CFA(Sigma)。在研究的第 18 天,根据体重将小鼠随机分组。入组后开始治疗,并持续每日一次(每24小时一次),直至研究第33天。动物经口服途径给予赋形剂(20%羟丙基-β-环糊精水溶液)或不同剂量的evobrutinib,或参考化合物甲氨蝶呤(MTX;0.5 mg/kg)。研究于第34天终止。在关节炎发作的第18-34天,使用以下标准对每只爪子(右前爪、左前爪、右后爪和左后爪)进行每日临床评分:0 = 正常;1 = 一个后爪或前爪关节受累或轻微弥漫性红斑和肿胀;2 = 两个后爪或前爪关节受累或轻度弥漫性红斑和肿胀;3 = 三个后爪或前爪关节受累或中度弥漫性红斑和肿胀;4 = 四个后爪或前爪关节受累或显著弥漫性红斑和肿胀; 5 = 整个爪子受累,出现严重的弥漫性红斑和严重肿胀,且无法屈曲趾部。对小鼠的前爪、后爪和膝关节进行组织病理学评分。炎症、血管翳形成、软骨损伤和骨吸收分别进行评分。将每个踝关节或膝关节的四个参数评分相加。计算每只动物所有六个组织的平均评分,并显示各组的平均评分。[3] SLE 模型[3] 10 周龄雌性 NZB/W F1 小鼠在第 0 天和第 1 天分别接受两次静脉注射,每次注射 1 × 10⁸ IU/100 μl 含有 mmIfna5_v1 插入片段的腺病毒(溶于生理盐水),或不进行任何处理(假手术)。在注射含有 mmIfna5_v1 插入片段的腺病毒 2 周后开始药物治疗,并持续到实验结束(10 周)。每组10只小鼠每日一次经口灌胃给予指定剂量的evobrutinib或300 mg/kg的吗替麦考酚酯。在指定日期采集血清和尿液样本,分别用于抗dsDNA抗体(ELISA法)和尿蛋白肌酐比值(UPCR;ADVIA 1800全自动生化分析仪测定)的测定。蛋白尿定义为UPCR > 3。此外,在实验最后一天,使用ADVIA1800全自动生化分析仪分析血清中的临床化学参数,包括尿素氮、白蛋白、总蛋白和胆固醇。在实验最后一天,使用Sysmex XT-2000iV血液分析仪对全血进行血液学分析。在实验最后一天,使用流式细胞仪分析脾细胞中的B细胞和T细胞亚群。图 9 显示了门控策略。脾细胞中 BTK 占有率的测定方法如 Honigberg 等人所述 小鼠 EAE 模型:用 MOG35-55 肽免疫 6-8 周龄 C57BL/6 小鼠以诱导 EAE。将小鼠随机分为载体组和治疗组。Evobrutinib (M2951) 溶于 10% DMSO + 90% 玉米油中,从免疫后第 0 天开始,以 10 或 30 mg/kg/天的剂量口服给药。每日评估临床评分;于第21天收集脊髓进行组织病理学分析(H&E染色和卢克索尔快蓝染色)[3] - 大鼠B细胞活化模型:雄性Wistar大鼠(200-250 g)在静脉注射抗IgM抗体(5 mg/kg)前1小时口服Evobrutinib (M2951)(10、30 mg/kg)。48小时后分离脾细胞进行B细胞增殖试验(MTT);采用ELISA法检测血清IgM水平[1] - 小鼠CIA模型:DBA/1小鼠用牛II型胶原蛋白免疫以诱导关节炎。从免疫后第14天开始,连续21天口服Evobrutinib (M2951)(30 mg/kg/天)。每3天测量一次爪体积;收集关节组织进行组织病理学评分[3] |
| 药代性质 (ADME/PK) |
小鼠体内BTK占有率的PK/PD模型[3]
本研究建立了临床前疾病模型中药物暴露量、靶点占有率和疗效之间的关系。采用一级吸收的双室模型估算了雄性DBA/1OlaHsd小鼠和雌性NZB/W小鼠的PK参数,分别用于RA和SLE模型。不同品系小鼠的PK参数见补充表II。相应的暴露量数据见补充图1F和1G。 一旦evobrutinib与BTK结合,其药效主要取决于体内BTK的再合成和降解速率,而非evobrutinib的全身暴露量。这是由于evobrutinib与BTK的共价结合所致,如图12A所示。利用图4C和4D所示的BTK占有率和暴露数据,对雄性DBA/1OlaHsd小鼠进行了BTK占有率PK/PD建模。该品系的PK参数用于参数化描述evobrutinib与BTK结合的二级速率常数kirrev。口服evobrutinib后,血浆浓度和BTK占有率的时间进程拟合到Abelö等人改进的PK/PD模型,该模型示意图如图12A所示。所选的PD模型包含一个间接反应模型,该模型同时描述了描述evobrutinib与BTK不可逆结合的二级速率常数(kirrev)和BTK蛋白的小鼠降解速率(kdeg)。小鼠白细胞中估计的PK/PD参数见补充表II,占有率剂量-反应时间进程如图12B所示。这些药物(kirrev)和系统(kdeg)参数被整合到一个模型中,该模型描述了每日给药后稳态下BTK占有率的波动。如图12C所示,该模型预测,小鼠每日服用1 mg/kg evobrutinib后,稳态下BTK占有率最高可达70%,最低可达50%。每日服用5 mg/kg时,BTK占有率最高可达97%,最低可达70%。预计更高剂量的evobrutinib对小鼠BTK占有率的时间进程影响甚微。这与体内 BTK 蛋白的周转而非 evobrutinib 的暴露量完全驱动 PD 效应相符。 口服生物利用度:72%(人),68%(大鼠),75%(犬)[2] - 血浆半衰期 (t1/2):8.5 小时(人),6.2 小时(大鼠),10.1 小时(犬)[2] - 血浆峰浓度 (Cmax):1.8 μg/mL(人,口服 50 mg),2.3 μg/mL(大鼠,口服 30 mg/kg)[2] - 分布容积 (Vd):1.3 L/kg(人),1.8 L/kg(大鼠)[2] - 清除率 (CL):0.15 L/h/kg(人),0.22 L/h/kg(大鼠)[2] - 代谢:主要经 CYP3A4 代谢;主要代谢产物为非活性代谢物 (M1) [2] - 排泄:约 65% 经粪便(以代谢物形式)排泄,约 25% 经尿液(以代谢物形式)排泄;原药 < 4% [2] |
| 毒性/毒理 (Toxicokinetics/TK) |
急性毒性:LD50 > 200 mg/kg(大鼠和小鼠口服)[1]
- 亚慢性毒性:大鼠每日口服 30 mg/kg,持续 28 天,未引起肝肾功能(ALT、AST、肌酐)或血液学参数的显著变化[1] - 血浆蛋白结合率:~98%(人),~97%(大鼠),~96%(犬)[2] - 临床不良事件:人体试验中出现轻度至中度腹泻(12%)、头痛(10%)和疲劳(8%);未见严重血液学或器官毒性[2] - 药物相互作用:CYP3A4 的弱抑制剂;与 CYP3A4 底物合用可能增加其血浆浓度[2] |
| 参考文献 | |
| 其他信息 |
Evobrutinib 正在临床试验 NCT03934502(膳食组成和给药时间对 Evobrutinib 生物利用度的影响)中进行研究。
Evobrutinib 是一种布鲁顿酪氨酸激酶 (BTK) 抑制剂,具有潜在的抗肿瘤活性。给药后,Evobrutinib 可抑制 BTK 的活性,并阻止 B 细胞抗原受体 (BCR) 信号通路的激活。这既能阻止 B 细胞活化,又能阻止 BTK 介导的下游生存通路的激活,从而抑制过度表达 BTK 的恶性 B 细胞的生长。BTK 是 Src 相关 BTK/Tec 家族胞质酪氨酸激酶的成员,在 B 细胞恶性肿瘤中过度表达;它在B淋巴细胞的发育、活化、信号传导、增殖和存活中发挥着重要作用。 药物适应症 治疗多发性硬化症 Evobrutinib (M2951)是一种强效、选择性、口服有效的共价BTK抑制剂[1, 2, 3] - 核心作用机制:与BTK Cys481共价结合,不可逆地抑制BTK活性,阻断BCR介导的信号传导,并抑制参与自身免疫反应的B细胞、单核细胞和T细胞的活化[1, 3] - 潜在的治疗应用:自身免疫性疾病,包括多发性硬化症(MS)、类风湿性关节炎(RA)和系统性红斑狼疮(SLE)[3] - 具有口服生物利用度高、半衰期长和选择性好的特点,支持每日一次给药剂量[2] - 临床开发阶段:在复发缓解型多发性硬化症的 III 期试验中进行了评估;已证实可有效降低复发率和 MRI 病灶负荷[3] |
| 分子式 |
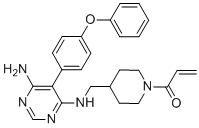
C25H27N5O2
|
|---|---|
| 分子量 |
429.5142
|
| 精确质量 |
429.216
|
| 元素分析 |
C, 69.91; H, 6.34; N, 16.31; O, 7.45
|
| CAS号 |
1415823-73-2
|
| 相关CAS号 |
1415823-73-2
|
| PubChem CID |
71479709
|
| 外观&性状 |
White to off-white solid powder
|
| 密度 |
1.2±0.1 g/cm3
|
| 沸点 |
683.4±55.0 °C at 760 mmHg
|
| 闪点 |
367.1±31.5 °C
|
| 蒸汽压 |
0.0±2.1 mmHg at 25°C
|
| 折射率 |
1.637
|
| LogP |
3.19
|
| tPSA |
93.4
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
32
|
| 分子复杂度/Complexity |
595
|
| 定义原子立体中心数目 |
0
|
| SMILES |
O=C(C([H])=C([H])[H])N1C([H])([H])C([H])([H])C([H])(C([H])([H])N([H])C2C(=C(N([H])[H])N=C([H])N=2)C2C([H])=C([H])C(=C([H])C=2[H])OC2C([H])=C([H])C([H])=C([H])C=2[H])C([H])([H])C1([H])[H]
|
| InChi Key |
QUIWHXQETADMGN-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C25H27N5O2/c1-2-22(31)30-14-12-18(13-15-30)16-27-25-23(24(26)28-17-29-25)19-8-10-21(11-9-19)32-20-6-4-3-5-7-20/h2-11,17-18H,1,12-16H2,(H3,26,27,28,29)
|
| 化学名 |
1-[4-[[[6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl]amino]methyl]piperidin-1-yl]prop-2-en-1-one
|
| 别名 |
MSC-2364447-C; MSC-2364447 C; Evobrutinib; 1415823-73-2; 1-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)piperidin-1-yl)prop-2-en-1-one; Evobrutinib [INN]; M-2951; M 2951; M2951; MSC-2364447C; MSC 2364447C; MSC2364447C
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ~86 mg/mL (~200.2 mM)
Ethanol: ~10 mg/mL (~23.3 mM) |
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3282 mL | 11.6412 mL | 23.2823 mL | |
| 5 mM | 0.4656 mL | 2.3282 mL | 4.6565 mL | |
| 10 mM | 0.2328 mL | 1.1641 mL | 2.3282 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Study of Evobrutinib in Participants With Relapsing Multiple Sclerosis (RMS)
CTID: NCT04032158
Phase: Phase 3 Status: Termi
A Phase II, Randomized, Double-Blind, Placebo-Controlled Dose-Ranging Study To Evaluate the Safety and Efficacy of M2951 in Subjects with Systemic Lupus Erythematosus (SLE)
CTID: null
Phase: Phase 2 Status: Completed, Prematurely Ended
Date: 2017-04-27
")
Figure 1. X-ray structure of BTK ligandB43bound to the BTK kinase domain.J Med Chem.2019 Aug 15. doi: 10.1021/acs.jmedchem.9b00794. |
|---|
") Figure 2. Overlay of crystal structures ofA5andA7.
Figure 5. Crystal structure of evobrutinib bound to the BTK kinase domain.J Med Chem.2019 Aug 15. doi: 10.1021/acs.jmedchem.9b00794. |
") Figure 3. PK/PD studies in mice.
Figure 6. Rat CIA model: rats treated with evobrutinib, MTX, or vehicle.J Med Chem.2019 Aug 15. doi: 10.1021/acs.jmedchem.9b00794. |
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
")
")
")
")
 463611831
463611831
