| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
CDK1 ≤1 μM (IC50) CDK2 ≤1 μM (IC50) CDK5 ≤1 μM (IC50) CDK7 ≤1 μM (IC50) CDK9 ≤1 μM (IC50)
|
|---|---|
| 体外研究 (In Vitro) |
缺血性中风是全球第二大死亡原因。缺血性卒中后,神经血管单位(NVU)炎症和外周白细胞浸润是导致脑损伤扩大的主要因素。长期以来,仅限于神经元,过去10年来,越来越多的研究集中在细胞周期蛋白依赖激酶(CDKs)对NVU其他细胞以及白细胞的作用上。在全脑和局灶性脑缺血模型中,使用最广泛的CDKs抑制剂(R)-罗斯科维汀及其(S)异构体都能减少脑损伤。我们之前的研究表明,(S)-roscovitine至少通过调节NVU对缺血的反应起作用。有趣的是,在几种炎症模型中,roscovitine被证明可以减少白细胞介导的炎症。roscovitine主要靶向CDK 1、2、5、7和9的特异性抑制表明,这些CDK在脑损伤(包括缺血性卒中)后NVU细胞和白细胞的炎症过程中起着关键作用。这里总结的数据支持了将罗司卡尼汀作为治疗缺血性卒中的潜在治疗剂的研究,并概述了脑缺血期间CDK 1、2、5、7和9在脑细胞和白细胞中的功能[1]。
|
| 体内研究 (In Vivo) |
(S)-Roscovitine(25 mg/kg;腹腔注射;pMCAo 前 15 分钟和 pMCAo 后 1 小时)在永久性大脑中动脉闭塞成年小鼠模型中表现出神经保护功效 [2]。
|
| 酶活实验 |
免疫沉淀和蛋白激酶测定[2]
将脑裂解物(500µg)与0.8µg p35抗体一起孵育。CDK5/p35和CDK5/p25免疫复合物用珠缓冲液(50 mM Tris pH 7.4、5 mM NaF、250 mM NaCl、5 mM EDTA、5 mM EGTA、0.1%NP-40、10µg/ml亮肽、抑肽酶和大豆胰蛋白酶抑制剂以及100µM苯甲脒)洗涤4次,用缓冲液C(60 mMβ-甘油磷酸、30 mM对硝基苯磷酸、25 mM Mops(pH 7.0)、5 mM EGFA、15 mM MgCl2、1 mM DTT、0.1 mM钒酸钠)洗涤1次。在最终体积为30µl的15µM[γ-33P]ATP(3000 Ci/mmol;10 mCi/ml)存在下,直接在缓冲液C中的珠粒上评估CDK5激酶活性,其中组蛋白H1/ml为1 mg。在30°C下孵育20分钟后,将20µl等分上清液点在2.5×3 cm的Whatman P81磷酸纤维素纸上,20秒后,在10 ml磷酸/升水溶液中洗涤过滤器四次(每次至少5分钟)。在1ml ACS闪烁液存在下对湿滤器进行计数。 |
| 动物实验 |
Animal/Disease Models: 20-25 g, P60 male C57 b/6 mice[2]
Doses: 25 mg/kg Route of Administration: I.p.; at 15 min before and 1 hr after pMCAo Experimental Results: Decreased of the total infarct volume. Blood brain barrier permeability [2] SD adult rats received intravenous injection of (S)-roscovitine solution (HPbCD 30% pH 7.4) at 2 doses (20 and 30 mg/kg; n = 2 rats/dose). Dosed animals were sacrificed at 15 min after the end of the intravenous injection. Blood were collected through the retro-orbital sinus using a capillary tube. Animals were then perfused with saline solution into the heart to extract the maximum blood sample from the brain. Brain was collected, and homogenized. Both plasma and brain samples were analyzed using LC/MS/MS determination, according to ADME-Bioanalysis procedures. Schemes of the experimental procedures are shown in Figures 2A, C . For experiments done at Neurokin ( Figure 2A ), (S)-roscovitine (n = 16 rats) or vehicle (n = 18 rats) was injected into the animals through a IV bolus (25 mg/kg in HPbCD 30%, pH 7.4) followed by 3 successive SC injections (54 mg/kg in saline HCl 50 mM, pH 1.5) performed 15 min before and at 24 and 29 hrs post-occlusion. For experiments performed at MDS Pharma Services ( Figure 2C ), drug (n = 11–12 rats) or vehicle (n = 13 rats) was administered through a bolus IV (25 mg/kg in HPbCD 30%, pH 7.4) followed by immediate SC infusion for 48 hrs, and performed either at 15 min prior or 2 hrs 15 min after the occlusion. Several drug doses were tested for the SC infusion (10, 5, and 1 mg/kg in saline HCl 50 mM, pH 1.5). Animal treatments were performed in a blind manner in both tMCAo studies. E18 mixed hippocampal cells, pharmacological treatments and evaluation of neuronal death [2] Mixed hippocampal cell cultures were prepared from embryonic day 18 (E18) Wistar rats as previously described and grown in vitro for 10 days. Treatments were done by directly adding KA (200 µM), drugs, and/or vehicle (DMSO 0.1%) to the medium and left for 5 hrs. No significant KA-induced toxic effect was ever detected on glial cells at this concentration (date not shown). The effects of roscovitine compounds [(R)-, (S)-, N6-methyl-(R)-, or O6-(R)-roscovitine] were determined by adding different concentrations of the molecules ranging from 0.05 to 50 µM to the medium at the same time as the KA addition. The cell death marker, propidium iodide (PI; 7.5 µM), was added to the medium at 4 hrs. Neuronal death was evaluated at 5 hrs by combining phase contrast and fluorescent microscopy observations. Neurons from random and representative fields were counted at low magnification (4x). At least 5 fields per condition (number total of neurons exceeding in general 150) were examined from 3 independent cultures. For every condition in every experiment, percentage of neuronal death was expressed as the ratio between PI-positive neurons and the total number of neurons visualized by phase contrast microscopy. To assess neuroprotection, relative neuronal death (RND) was calculated in 3 independent cultures and a neuroprotection index (NI) defined as: RND = (% of neuronal death with KA/roscovitine – % of neuronal death with roscovitine)/(% of neuronal death with KA - % of neuronal death with vehicle), and NI = 1-RND. By definition, relative percentage of neuronal death (RND) in KA-treated culture was 100% and neuroprotection index (NI) was 0%. |
| 参考文献 |
|
| 其他信息 |
(S)-Roscovitine and Infarct Size in Focal Ischemia [1]
(S)-roscovitine ICV administration to mice 48 h before pMCAo and throughout the duration of pMCAo led to a 28% decrease of infarct volume compared to vehicle-treated animals at 3 h post-occlusion. Systemic administration of (S)-roscovitine by two successive IP injections at 15 min prior and 1 h after the occlusion led to a 31% decrease of the total infarct volume at 3 h post-occlusion, showing no loss of neuroprotective effect. For both administration modes, the hypometabolic zone volume, but not the infarct core, decreased in (S)-roscovitine-treated animals compared to vehicle. Interestingly, they observed that the increase of CDK5 activity post-pMCAo was prevented by (S)-roscovitine treatment, suggesting that the beneficial effect of (S)-roscovitine was at least partly due to CDK5 inhibition. (S)-roscovitine neuroprotective efficacy was also assessed on two independents blinded studies in a tMCAo rat model. In the first study, a 90 min tMCAo rat model, (S)-roscovitine was administered by IV bolus 15 min prior to ischemia followed by three successive SC injections at 15 min prior to and 24 h and 29 h after the occlusion. (S)-roscovitine significantly decreased the infarct volume by 30% 48 h after reperfusion. In the second study, a 120 min tMCAo rat model, (S)-roscovitine was administered by IV bolus followed by continuous SC infusion performed 135 min after (post-MCA) the occlusion, leading to a significant decrease by 27% of the infarct volume. Rousselet et al. studied (S)-roscovitine effect in a randomized blind study on a tMCAo rat model. (S)-roscovitine administration 15 min post-reperfusion by IV bolus followed by 48 h SC infusion decreased infarct volume by 21%, 48 h after reperfusion. [1] Background: Although quite challenging, neuroprotective therapies in ischemic stroke remain an interesting strategy to counter mechanisms of ischemic injury and reduce brain tissue damage. Among potential neuroprotective drug, cyclin-dependent kinases (CDK) inhibitors represent interesting therapeutic candidates. Increasing evidence indisputably links cell cycle CDKs and CDK5 to the pathogenesis of stroke. Although recent studies have demonstrated promising neuroprotective efficacies of pharmacological CDK inhibitors in related animal models, none of them were however clinically relevant to human treatment. Methodology/principal findings: In the present study, we report that systemic delivery of (S)-roscovitine, a well known inhibitor of mitotic CDKs and CDK5, was neuroprotective in a dose-dependent manner in two models of focal ischemia, as recommended by STAIR guidelines. We show that (S)-roscovitine was able to cross the blood brain barrier. (S)-roscovitine significant in vivo positive effect remained when the compound was systemically administered 2 hrs after the insult. Moreover, we validate one of (S)-roscovitine in vivo target after ischemia. Cerebral increase of CDK5/p25 activity was observed 3 hrs after the insult and prevented by systemic (S)-roscovitine administration. Our results show therefore that roscovitine protects in vivo neurons possibly through CDK5 dependent mechanisms. [2] |
| 分子式 |
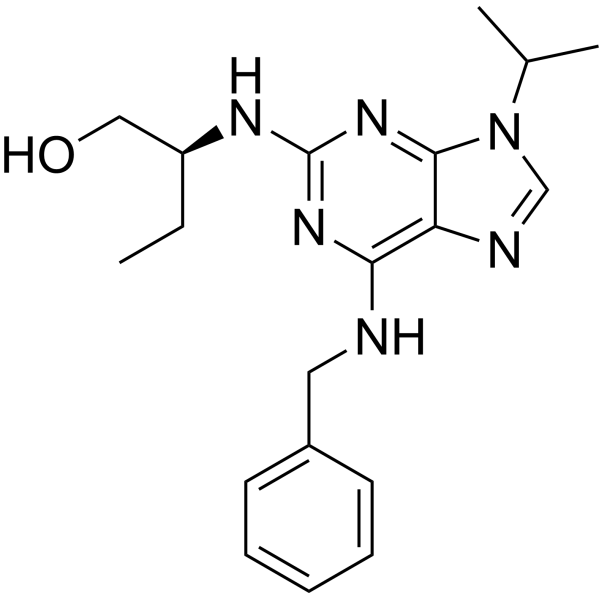
C19H26N6O
|
|---|---|
| 分子量 |
354.44934
|
| 精确质量 |
354.216
|
| 元素分析 |
C, 64.38; H, 7.39; N, 23.71; O, 4.51
|
| CAS号 |
186692-45-5
|
| PubChem CID |
6603989
|
| 外观&性状 |
Typically exists as solids at room temperature
|
| LogP |
3.2
|
| tPSA |
87.9 Ų
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
26
|
| 分子复杂度/Complexity |
417
|
| 定义原子立体中心数目 |
1
|
| SMILES |
CC[C@H](NC1=NC(NCC2C=CC=CC=2)=C2N=CN(C(C)C)C2=N1)CO
|
| InChi Key |
BTIHMVBBUGXLCJ-HNNXBMFYSA-N
|
| InChi Code |
InChI=1S/C19H26N6O/c1-4-15(11-26)22-19-23-17(20-10-14-8-6-5-7-9-14)16-18(24-19)25(12-21-16)13(2)3/h5-9,12-13,15,26H,4,10-11H2,1-3H3,(H2,20,22,23,24)/t15-/m0/s1
|
| 化学名 |
(2S)-2-[[6-(benzylamino)-9-propan-2-ylpurin-2-yl]amino]butan-1-ol
|
| 别名 |
(S)-Seliciclib; (S)-ROSCOVITINE; 186692-45-5; (S)-Seliciclib; Seliciclib, (S)-; Seliciclib (2S)-form [MI]; (2S)-2-[[6-(benzylamino)-9-propan-2-ylpurin-2-yl]amino]butan-1-ol; UNII-8C43G94891; Roscovitine, (S)-Isomer; (S)-CYC202
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8213 mL | 14.1064 mL | 28.2127 mL | |
| 5 mM | 0.5643 mL | 2.8213 mL | 5.6425 mL | |
| 10 mM | 0.2821 mL | 1.4106 mL | 2.8213 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 CDK8-IN-16
CDK8-IN-16
 CDK2-IN-37
CDK2-IN-37
 CDK7-IN-32
CDK7-IN-32
 CDK-IN-16
CDK-IN-16
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
