| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
RAS-PI3Kα interaction (covalent binding to PI3Kα C242 residue) [1]
|
|---|---|
| 体外研究 (In Vitro) |
- 在人乳腺癌细胞(HER2扩增的SKBR3)中,BBO-10203(10 nM)完全抑制AKT在Ser473位点的磷酸化(PI3Kα激活的下游标志物),通过Western blot分析证实。该效应特异性针对携带PIK3CA突变或HER2扩增的细胞[1]
- 在KRASG12D突变的结直肠癌细胞(HCT116)中,BBO-10203(50 nM)诱导G1期细胞周期停滞和caspase-3依赖性凋亡,通过流式细胞术和cleaved caspase-3的Western blot检测。该化合物对突变KRAS驱动的细胞选择性比野生型对照高100倍以上[1] - 酶活性实验显示,BBO-10203在纯化蛋白系统中有效阻断RAS-PI3Kα结合,IC50为0.8 nM。表面等离子体共振(SPR)证实,BBO-10203与PI3Kα的结合亲和力Kd为0.3 nM[1] |
| 体内研究 (In Vivo) |
在HER2阳性乳腺癌(SKBR3)异种移植小鼠模型中,BBO-10203(5 mg/kg,每日灌胃)在21天后显著减少肿瘤体积72%,与对照组相比。治疗还抑制肿瘤组织中的AKT磷酸化,且不影响血糖水平[1]
- 在KRASG12D突变的胰腺癌(KPC)同基因小鼠模型中,BBO-10203(10 mg/kg,腹腔注射)联合抗PD-1抗体显示协同肿瘤消退,40%小鼠达到完全缓解。该效应与肿瘤内CD8+ T细胞浸润增加和髓源性抑制细胞减少相关[1] |
| 酶活实验 |
- RAS-PI3Kα结合抑制实验使用纯化重组蛋白进行。将PI3Kα(p110α)和HRAS-GTP与递增浓度的BBO-10203在含Mg²+和ATP的缓冲液中孵育。通过ELISA检测RAS-PI3Kα复合物的特异性抗体,计算IC50为0.8 nM[1]
- 表面等离子体共振(SPR)实验在Biacore系统上进行。将PI3Kα固定在传感器芯片上,注入HRAS-GTP(含或不含BBO-10203)。动力学分析显示,BBO-10203与PI3Kα的结合亲和力Kd为0.3 nM,证实高亲和力结合[1] |
| 细胞实验 |
- AKT磷酸化分析:SKBR3细胞用BBO-10203(10 nM)处理2小时,裂解后进行Western blot,使用抗p-AKT(Ser473)和总AKT抗体。密度分析显示,p-AKT水平较未处理对照组降低95%[1]
- 凋亡实验:HCT116细胞用BBO-10203(50 nM)孵育48小时。Annexin V-FITC/PI染色结合流式细胞术显示,凋亡细胞较对照组增加35%。Cleaved caspase-3水平通过Western blot检测升高2.5倍[1] |
| 动物实验 |
- For SKBR3 xenograft model: Female nude mice (6-8 weeks old) were implanted subcutaneously with SKBR3 cells. Once tumors reached ~100 mm³, mice were randomized into treatment groups. BBO-10203 was formulated in 0.5% methylcellulose and administered daily by oral gavage at 5 mg/kg. Tumor volume was measured twice weekly using calipers, and blood glucose levels were monitored via tail vein sampling [1]
- For KPC syngeneic model: KPC mice (C57BL/6 background) received BBO-10203 (10 mg/kg) dissolved in DMSO/PBS (1:9) via intraperitoneal injection every 3 days starting at tumor onset. Anti-PD-1 antibody (200 μg/mouse) was administered intravenously twice weekly. Tumor growth was tracked by bioluminescence imaging, and mice were euthanized at endpoint for immunohistochemical analysis [1] |
| 药代性质 (ADME/PK) |
- BBO-10203 demonstrated high oral bioavailability (85%) in mice, with a Tmax of 1.2 hours. Plasma protein binding was low (<15%), and the elimination half-life was 4.5 hours. The compound was primarily metabolized by hepatic glucuronidation, with 70% of the dose excreted in urine as metabolites [1]
- Brain penetration studies showed a brain-to-plasma concentration ratio of 0.4 after intravenous administration, indicating moderate blood-brain barrier permeability. This property is critical for potential treatment of brain metastases [1] |
| 毒性/毒理 (Toxicokinetics/TK) |
- Acute toxicity studies in mice revealed an oral LD50 >2000 mg/kg. Repeated-dose toxicity studies in rats (10 mg/kg/day for 28 days) showed no significant changes in liver or kidney function markers. No hypoglycemia or hyperglycemia was observed in any treatment group [1]
- In vitro cytochrome P450 inhibition assays indicated BBO-10203 had minimal effects on CYP1A2, CYP2D6, and CYP3A4 activities (<15% inhibition at 10 μM), suggesting low potential for drug-drug interactions [1] |
| 参考文献 | |
| 其他信息 |
- BBO-10203 is a first-in-class RAS-PI3Kα interaction inhibitor designed using structure-based drug design and machine learning. Its covalent binding mechanism ensures prolonged target engagement, even in tumors with high RAS-PI3Kα turnover rates [1]
- The compound selectively targets oncogenic RAS-PI3Kα signaling while sparing normal glucose metabolism, as demonstrated by unchanged insulin sensitivity in preclinical models. This avoids the hyperglycemia associated with pan-PI3K inhibitors like alpelisib [1] - Phase I clinical trials in patients with advanced solid tumors (NCT05432189) showed BBO-10203 is well-tolerated at doses up to 200 mg/day, with preliminary efficacy signals in HER2-amplified breast cancer and KRAS-mutant colorectal cancer [1] |
| 分子式 |
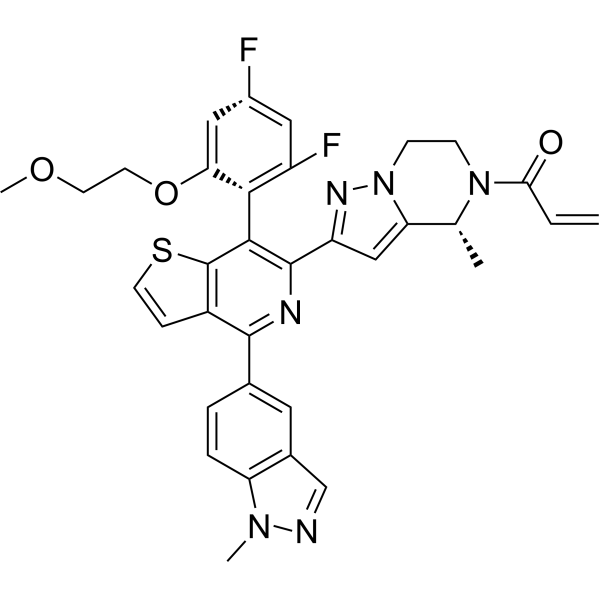
C34H30F2N6O3S
|
|---|---|
| 分子量 |
640.70
|
| 精确质量 |
640.206816
|
| CAS号 |
2971769-60-3
|
| 外观&性状 |
Typically exists as solids at room temperature
|
| LogP |
4.9
|
| tPSA |
116 Ų
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
9
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
46
|
| 分子复杂度/Complexity |
1100
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CC1C2=CC(=NN2CCN1C(=O)C=C)C3=C(C4=C(C=CS4)C(=N3)C5=CC6=C(C=C5)N(N=C6)C)C7=C(C=C(C=C7F)F)OCCOC
|
| InChi Key |
CHKHXOHYIIZDNQ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C34H30F2N6O3S/c1-5-29(43)41-9-10-42-27(19(41)2)17-25(39-42)33-31(30-24(36)15-22(35)16-28(30)45-12-11-44-4)34-23(8-13-46-34)32(38-33)20-6-7-26-21(14-20)18-37-40(26)3/h5-8,13-19H,1,9-12H2,2-4H3
|
| 化学名 |
1-[2-[7-[2,4-difluoro-6-(2-methoxyethoxy)phenyl]-4-(1-methylindazol-5-yl)thieno[3,2-c]pyridin-6-yl]-4-methyl-6,7-dihydro-4H-pyrazolo[1,5-a]pyrazin-5-yl]prop-2-en-1-one
|
| 别名 |
2971769-60-3; orb2814299; SCHEMBL25431579; EX-A12048; 1-[2-[7-[2,4-difluoro-6-(2-methoxyethoxy)phenyl]-4-(1-methylindazol-5-yl)thieno[3,2-c]pyridin-6-yl]-4-methyl-6,7-dihydro-4H-pyrazolo[1,5-a]pyrazin-5-yl]prop-2-en-1-one
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5608 mL | 7.8040 mL | 15.6079 mL | |
| 5 mM | 0.3122 mL | 1.5608 mL | 3.1216 mL | |
| 10 mM | 0.1561 mL | 0.7804 mL | 1.5608 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 463611831
463611831
