| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
Histamine H4 receptor/H4 receptor (pKi = 7.94)
|
|---|---|
| 体外研究 (In Vitro) |
Maleate ST-1006 (10 μM) 是一种强大的嗜碱性粒细胞迁移诱导剂,可引起嗜碱性粒细胞迁移[2]。 ST-1006 (0-100 μM)maleate 抑制 FceRI 介导的嗜碱性粒细胞活化,并降低 FceRI 触发的嗜碱性粒细胞上 CD63 和 CD203c 的表达水平[2]。
我们可以证明,高纯度的嗜碱性细胞表达H1R、H2R和H4R,但不表达H3R mRNA。人嗜碱性细胞H4R mRNA表达水平高于H1R表达水平(P < 0.01)。组胺和H4R激动剂ST-1006可启动嗜碱性细胞的主动迁移(P < 0.001)。用组胺或特异性H4R激动剂ST-1006预孵育后,细胞表面CD63和CD203c的表达显著降低(P < 0.01)。组胺或H4R激动剂ST-1006均能显著降低嗜碱性细胞在不同刺激下(fc - ri交联或膜翅目蜂毒过敏原刺激)活化后硫代白三烯的合成和释放(P < 0.05 ~ 0.001)。 结论:这些数据表明,H4R调节人类嗜碱性细胞中ige依赖的过程,并提供了H4R通过参与负反馈回路来防止压倒性免疫反应的新功能。[2] |
| 体内研究 (In Vivo) |
ST-1006(1-100 mg/kg;皮下;雄性 CD-1 小鼠)马来酸盐具有止痒和抗炎特性[1]。
组胺H4受体(H4R)拮抗剂的抗炎作用为炎症/变应性疾病的治疗开辟了新的治疗选择,但H4R在炎症中的作用远未得到解决。本研究旨在探讨不同功效和功能的氨基嘧啶类结构相关的H4R配体(中性拮抗剂ST-994、部分激动剂ST-1006、逆激动剂ST-1012和部分逆激动剂ST-1124)对巴丁油致小鼠耳部水肿和瘙痒的作用。H4R配体在外用巴豆油前皮下注射。虽然ST-1006和ST-1124在任何剂量(10-100 mg/kg)下均无效,但ST-994和ST-1012(30和100 mg/kg)均可显著减轻巴豆油诱导的耳部水肿。此外,ST-994、ST-1006和ST-1124在30 mg/kg时对巴豆油致耳部瘙痒有显著抑制作用,而ST-1012则无显著抑制作用。根据参考H4R拮抗剂JNJ7777120 (100 mg/kg)的结果,炎症耳组织的组织学检查表明,ST-994 (30 mg/kg)治疗导致炎症严重程度评分和浸润组织的嗜酸粒细胞数量显著降低,而炎症组织中脱颗粒肥大细胞的数量比完整肥大细胞的数量增加。这些数据表明,巴豆油引起的耳部炎症和瘙痒似乎明显但不同地受到H4R配体的影响。H4R中性拮抗剂ST-994作为一种治疗炎症性疾病的新方法,其双重作用的潜在优势必须仔细考虑。 |
| 细胞实验 |
嗜碱性粒细胞活化试验[2]
采用多色流式细胞术检测嗜碱性细胞活化标志物CD63和CD203c的表面表达。简单地说,将健康受试者的100 μl全血稀释在100 μl由PBS加IL-3 (2 ng/ml)组成的刺激缓冲液中。细胞在37℃下仅用刺激缓冲液、组胺(10 μM)或H4R激动剂ST-1006 (10 μM)预孵育15 min。剂量动力学实验用不同浓度的组胺或H4R激动剂(0.1 ~ 100 μM)刺激细胞。在时间动力学实验中,细胞分别与组胺或H4R激动剂预孵育15、10、5、0分钟,然后加入抗ige抗体20分钟。为了证明这些作用对H4R具有特异性,在组胺或H4R刺激之前,将细胞与H4R拮抗剂JNJ7777120预孵育30分钟。然后,加入与人IgE ε-链结合的单克隆抗人IgE抗体,活化细胞20 min。用fitc偶联抗cd63、植红蛋白偶联抗cd203c和过磷酸酶偶联抗fcε ri对细胞进行染色。通过裂解反应去除红细胞,流式细胞术对细胞进行分析。 硫代铕三烯ELISA试剂盒[2] 市购的细胞抗原刺激试验ELISA用于量化分离的白细胞在接触过敏原或刺激促进脱颗粒时产生的硫代白三烯(包括白三烯C4 (LTC4)及其衍生物LTD4和LTE4)的释放。白细胞分离、细胞刺激和白三烯测定按照供应商的方案进行。对有过敏史的患者或膜翅目毒液致敏的患者采集全血。简而言之,用葡聚糖密度梯度离心法从全血中分离白细胞。将分离的白细胞重悬于含有IL-3的刺激缓冲液中。对每个样品进行了基础释放和不同刺激后的释放测试。为了激活嗜碱性粒细胞,在37°C下(i)用针对高亲和力IgE受体的抗体刺激细胞40分钟,(ii)用C5a (0.1 μM)、甲氧基-甲氧基-亮基-苯丙氨酸(FMLP) (1 μM)和血小板活化因子(PAF) (10 μM)的混合物刺激细胞。将细胞与蜂毒(200 μg/ml)在37℃下孵育40 min,以检测细胞对嗜碱性粒细胞的变应原依赖性脱颗粒作用。组胺(10 μM)、H4R激动剂ST-1006 (10 μM)、H1R激动剂2-吡啶乙胺(10 μM)和H2R激动剂胺胺(10 μM)随激活刺激同时加入。以不同浓度(0.1 ~ 100 μM)的H2R激动剂氨胺或H4R激动剂ST-1006刺激细胞,显示其浓度依赖性。在时间动力学实验中,细胞分别与H2R激动剂或H4R激动剂各10 μM分别预孵育10、5、0 min,然后加入蜂毒过敏原40 min,最后离心,收集上清。根据制造商的说明,使用CAST酶联免疫吸附测定法估计硫代溴三烯释放量。 |
| 动物实验 |
Animal/Disease Models: Male CD-1 mice with pruritus (8-10 weeks and 25-30 g)[1]
Doses: 1-100 mg/kg Route of Administration: subcutaneous (sc) injection Experimental Results: Had an antipruritic effect at the non-anti-inflammatory dose of 30 mg/kg. Edema [1] The technique originally described by Tubaro et al. (1986) and modified by Coruzzi et al. (2012) was followed. All experiments were carried out between 10 a.m. and 3 p.m., in order to avoid the influence of circadian variations in corticosteroid levels in the inflammatory responses. Cutaneous inflammation was induced in conscious mice by topical application of croton oil (2.5% in acetone). The irritant agent was applied with a micropipette (20 μl/ear) to the inner surface of the right ear. Acetone (20 μl/ear) was applied to the left ear, which served as a control (Tubaro et al. 1986). The H4R ligands ST-994, ST-1006, ST-1012, and ST-1124 (10–100 mg/kg); the reference H4R antagonist JNJ7777120 (30 and 100 mg/kg); or the vehicle were administered as single subcutaneous (s.c.) injection immediately before the topical application of croton oil. Control mice received s.c. injection with vehicle (20% DMSO and 80% 2-hydroxypropyl-β-cyclodextrin). Two or four hours after croton oil application, mice were sacrificed by cervical dislocation; both right (croton oil in acetone) and left (acetone) ears were removed by cutting horizontally across the indentation at the base of the ear. For each mouse, the extent of the edema was expressed as the difference in weight (Δ mg) between right (inflamed) and left (uninflamed) ear. Pruritus [1] As an indicator of pruritus, the scratching behavior of mice was determined (Fujii et al. 2010; Coruzzi et al. 2012). Single s.c. administration of vehicle, ST-994, ST-1006, ST-1012, or ST-1124 (30 mg/kg) was given immediately before croton oil application. Itch was measured by blinded counting of the number of scratching bouts in the 1-h period immediately following the application of croton oil. A bout of scratching was defined as three or more individual rapid scratch movements with the hind paw around the inflamed ear (Dunford et al. 2007). Hind paw movements directed away from the inflamed ear and grooming movements were not counted. Pruritus was evaluated as a difference in counting the scratching bouts between right (inflamed) and left (uninflamed) ear at 10-min intervals (time course, Δ scratching bouts/10 min) or as a difference in counting the total scratching bouts between right (inflamed) and left (uninflamed) ear over the 1-h observation period (Δ scratching bouts/1 h). Histology [1] For histological analysis, inflamed ears from vehicle-, ST-994-, ST-1006-, ST-1012-, ST-1124-, JNJ7777120-, or dexamethasone-treated CD-1 mice were removed 2 h after croton oil application, divided into two tissue samples, and fixed overnight in 10% formalin (Coruzzi et al. 2012). Subsequently, tissue samples were embedded longitudinally in paraffin, cut with a microtome into 4-μm sections, and mounted on glass slides. The cross-sections were stained with hematoxylin and eosin for the evaluation of inflammation intensity (total severity score) and the count of eosinophils. Each section was scored by two observers unaware of the treatment, according to the following score system: 0 = no inflammatory cells; 0.5 = rare leucocytes in the interstitium; 1 = rare leucocyte clusters infiltrating perivascular spaces (2–5 per section); 2 = leucocyte clusters infiltrating perivascular spaces (6–10 per section); and 3 = leucocyte clusters infiltrating the interstitium. The total severity score was then determined in the inflamed ear. The maximum severity score was six. Moreover, in the hematoxylin and eosin-stained cross-sections, the number of eosinophils was counted in ten randomly fields per inflamed ear and the total number was considered (number of eosinophils/ear). |
| 参考文献 |
|
| 其他信息 |
Results of the present study indicate that croton oil-induced ear inflammation and pruritus in CD-1 mice seem to be clearly, but variably, affected by the structurally related H4R ligands tested (Sander et al. 2009; Kottke et al. 2011; Gschwandtner et al. 2013). Subcutaneous injections of both the hH4R neutral antagonist ST-994 and the hH4R full inverse agonist ST-1012 inhibited croton oil-induced ear edema at any time point considered (2 and 4 h), while both the hH4R partial agonist ST-1006 and the hH4R partial inverse agonist ST-1124 were inactive. Croton oil-induced itch was not modified by ST-1012 at the anti-inflammatory dose of 30 mg/kg s.c., whereas the anti-inflammatory effect of ST-994 was associated to its antipruritic properties. Surprisingly, both ST-1006 and ST-1124 reduced croton oil-induced scratching bouts at a dose of 30 mg/kg s.c. that did not affect croton oil-induced ear edema, giving further evidence for the complex regulation behavior: (1) H4R blockade induced by ST-994 did not completely overcome results seen with the reference H4R antagonist JNJ7777120 as ST-994 reduced both croton oil-induced ear edema and pruritus while JNJ7777120 reduced inflammation but not scratching bouts (Coruzzi et al. 2012); (2) the anti-inflammatory effect of ST-994 is not explained by the antipruritic effect, since ST-1012 reduced croton oil-induced ear edema but not itch; (3) both ST-1006 and ST-1124 did not possess proinflammatory or anti-inflammatory effects in this model of acute inflammation when s.c. injected, but reductions of croton oil-induced pruritus indicate that their ineffectiveness depends on the parameter considered; and (4) differently from the hH4R neutral antagonist ST-994, the hH4R full inverse agonist ST-1012 (100 mg/kg s.c.) reduced only the ear edema developing at 2 h after challenge, whereas the late inflammatory response (at 4 h) was not affected; a possible explanation could be related to an unfavorable kinetics, but the effectiveness of ST-1012 (30 mg/kg s.c.) at 2 and 4 h after croton oil application minimizes this hypothesis. Taken together, the present and literature data suggest that the effects of H4R ligands are highly dependent on the strain (Coruzzi et al. 2012) and the specific model and/or parameter considered (present study). The anti-inflammatory effect of ST-994 or ST-1012 is in accordance with the H4R blockade in different settings in mice (Cowden et al. 2010; Coruzzi et al. 2012; Lucarini et al. 2016), but different from the ineffectiveness of JNJ7777120 on ear swelling induced by the two haptens, 2,4-dinitrochlorobenzene (Rossbach et al. 2008) or 2,4,6-trinitrochlorobenzene (Seike et al. 2010). Several factors may be responsible for these contrasting effects, including the type of chemical irritant, the presence/absence of a prior sensitization or the different cytokine profile involved, the use of different strains of mice and, in the same strain, the assay employed (Coruzzi et al. 2012) and the possibility of different pharmacokinetics of the different compounds.
At 2 h, both ST-994 and ST-1012 induced ear edema reductions as effectively as inhibitions induced by JNJ7777120 or the first-generation H1R blocker pyrilamine suggesting that both H1Rs and H4Rs are involved almost in the early phase of the inflammation. Even more interesting is the evidence that pyrilamine abolished croton oil-induced pruritus, while JNJ7777120 induced only a nonsignificant reduction (Coruzzi et al. 2012; Adami et al. 2014). In the same model of acute skin inflammation, the second-generation H1R antagonist cetirizine, that differs from first generation because of its high specificity and affinity for peripheral H1Rs, scarcely reduced ear edema and pruritus (Adami et al. 2014) thus opening the question if central penetration of the compound is necessary for both anti-inflammatory effect and/or antipruritic activity. Differences in the ability of antihistamines to interact with P-glycoprotein (P-gp) efflux pump at the blood-brain barrier (BBB) may determine their central nervous system (CNS) penetration (Chen et al. 2003; Obradovic et al. 2007) and as a consequence the presence or absence of central side effects. Sedating H1R antagonists are not P-gp substrates, while some non-sedating H1R antagonists are P-gp substrates: affinity for P-gp at BBB may explain the lack of CNS side effects of the second-generation H1R antagonists (Chen et al. 2003). The lack of effect on croton oil-induced ear edema by ST-1006 or ST-1124, together with their effectiveness in diminishing croton oil-induced itch, is surprising and even paradoxical for ST-1006 considering its pruritogen-evoked scratching after intradermal injection into the back skin of female BALBc mice (Rossbach et al. 2014). This paradoxical result is however in agreement with reductions of local reactions and pruritus in atopic dermatitis patients induced by intracutaneous injections of histamine (Heyer et al. 1998). Despite differences in strain can markedly influence scratching responses to histamine (Inagaki et al. 2001), it has to underline that the previously reported high susceptibility of NMRI mice to H4R-induced itching was not correlated to higher H4R densities in this mouse strain (Bäumer et al. 2008). Thus, any conclusion should be taken with caution because it may depend on the prototypical H4R ligand used, not neglecting its different efficacies in different cell/disease states (Gbahou et al. 2003; Sivertsen et al. 2013).[1] As life-threatening anaphylactic reactions to bee or wasp stings occur in 0.8–5% of the general population, we focussed in the main part of the study on the role of histamine and the H4R in this scenario, using tools which are used in the daily routine diagnostic of insect venom allergy. We could show the capability of histamine and the H4R agonist ST-1006 to suppress the FcεRI-induced expression of the activation markers CD63 and CD203c on the cell surface of basophils in a dose- and time-dependent manner by means of flow cytometry. Moreover, when using leukotriene secretion as an indicator of basophil degranulation, we could demonstrate that the synthesis and release of sulfidoleukotrienes from effector cells after activation with different stimuli or by FcεRI cross-linking were markedly reduced by selective stimulation with histamine and the H4R agonist ST-1006 at the same time period. In addition, we showed that the stimulation with histamine or the H4R agonist ST-1006 alone did not alter the basal degranulation in human basophils. These observations are consistent with Hofstra et al. who showed that also murine bone marrow mast cells were not affected by stimulation with histamine alone in terms of leukotriene and prostaglandin release. Several immunologic effects of histamine during basophil or mast cell activation in both in vitro assays and in the course of allergen immunotherapy were described in the literature. Former studies provided evidence that histamine is able to inhibit its own release in human leukocytes or mast cells. It was shown in one of these early studies that histamine and different H2R agonists inhibit the histamine release from human leukocytes with high potency. This effect was specifically blocked by an H2R antagonist, indicating that the H2R is involved 17. We could show the suppression of allergen-induced release of sulfidoleukotrienes from human basophils in the presence of histamine, the selective H2R agonist amthamine, or the selective H4R agonist ST-1006, confirming these early observations with regard to the H2R and expanding them to the H4R.[2] |
| 分子式 |
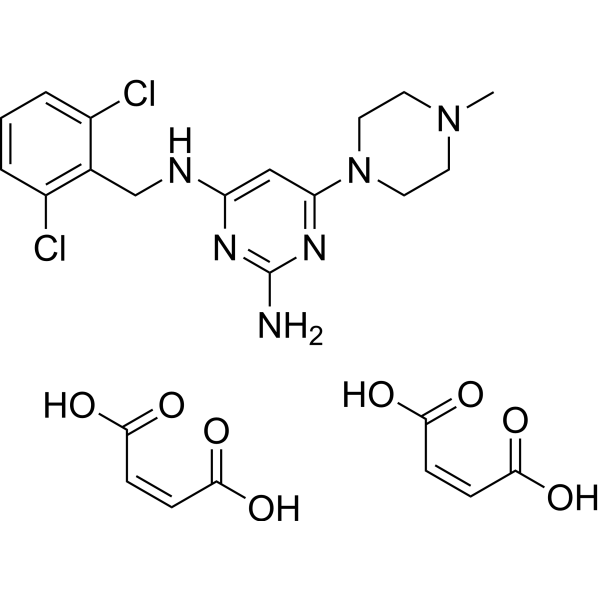
C24H28CL2N6O8
|
|---|---|
| 分子量 |
599.42
|
| 精确质量 |
598.134
|
| CAS号 |
1196994-12-3
|
| 相关CAS号 |
ST-1006;1196994-11-2
|
| PubChem CID |
165437225
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
220
|
| 氢键供体(HBD)数目 |
6
|
| 氢键受体(HBA)数目 |
14
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
40
|
| 分子复杂度/Complexity |
502
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CN1CCN(CC1)C2=NC(=NC(=C2)NCC3=C(C=CC=C3Cl)Cl)N.C(=C\C(=O)O)\C(=O)O.C(=C\C(=O)O)\C(=O)O
|
| InChi Key |
NBLGNUAWAKUPSY-SPIKMXEPSA-N
|
| InChi Code |
InChI=1S/C16H20Cl2N6.2C4H4O4/c1-23-5-7-24(8-6-23)15-9-14(21-16(19)22-15)20-10-11-12(17)3-2-4-13(11)18;2*5-3(6)1-2-4(7)8/h2-4,9H,5-8,10H2,1H3,(H3,19,20,21,22);2*1-2H,(H,5,6)(H,7,8)/b;2*2-1-
|
| 化学名 |
(Z)-but-2-enedioic acid;4-N-[(2,6-dichlorophenyl)methyl]-6-(4-methylpiperazin-1-yl)pyrimidine-2,4-diamine
|
| 别名 |
ST-1006 Maleate; ST-1006 (Maleate); 1196994-12-3;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.6683 mL | 8.3414 mL | 16.6828 mL | |
| 5 mM | 0.3337 mL | 1.6683 mL | 3.3366 mL | |
| 10 mM | 0.1668 mL | 0.8341 mL | 1.6683 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Granuliberin R
Granuliberin R
 Promethazine-d4 hydrochloride (Promethazine d4 hydrochloride)
Promethazine-d4 hydrochloride (Promethazine d4 hydrochloride)
 N-Desmethyl diphenhydramine-d3 hydrochloride
N-Desmethyl diphenhydramine-d3 hydrochloride
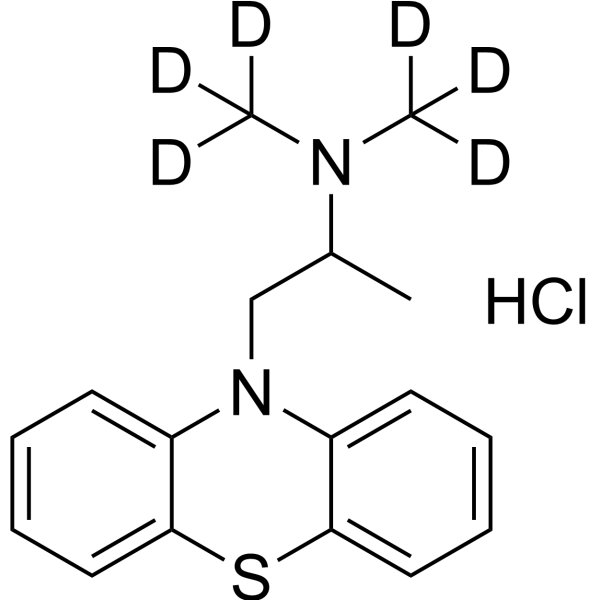 Promethazine-d6 hydrochloride ((±)-Promethazine-d6 (hydrochloride))
Promethazine-d6 hydrochloride ((±)-Promethazine-d6 (hydrochloride))
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1






























