| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
IC50: 25 nM (rat cortical neurons)[1]
|
|---|---|
| 体外研究 (In Vitro) |
CP-465022 (0.0001 μM-10 μM) 以非常缓慢的方式降低红藻氨酸诱导的反应,并且取决于化合物浓度,显示预测的 IC50 为 25 nM,在 3.2 μM 时几乎完全抑制[1]。 CP-465022 1 µM 持续 10 分钟不会对峰值 NMDA 感应电流产生显着影响,但在 NMDA 应用期间 8 秒测得的电流降低了 26%。在皮质和小脑颗粒神经元的原代培养物中,10 µM 的 CP-465,022 可抑制 NMDA 诱导的峰值电流 36%,并在 8 秒时测量的电流抑制 70%[1]。在培养的大鼠小脑颗粒神经元中,CP-465022 1 µM 持续 10 分钟可降低峰值 NMDA 电流,平均抑制 19%,并且在 8 秒时评估的 NMDA 电流降低 45%,这与在皮质神经元中观察到的结果相当[1] 。在电压钳位的大鼠海马切片中,CP-465022 (100 nM – 10 µM) 对红藻氨酸诱导的全细胞电流表现出抑制作用。在 100 nM 时,CP465,022 可抑制形成超过 200 秒的红藻氨酸电流,而在 500 nM 和 1 µM 时,CP-465,022 几乎完全抑制这些电流 (99.3%)[1]。
CP-465,022对皮质神经元AMPA受体介导电流的影响[1] 研究了CP-465022对红藻氨酸诱导的大鼠皮质神经元原代培养全细胞电流的影响。这种反应的药理学表明,这些红藻氨酸诱导的反应是由AMPA受体介导的。具体而言,(1)红藻氨酸的EC50(约145μM,见下文)与红藻氨酸作为低亲和力AMPA受体激动剂的作用一致(Partin等人,1993)。 如图2A所示,CP-465,022抑制了100μM红藻氨酸诱导的全细胞电流。抑制作用呈浓度依赖性,计算出的IC50为25 nM(图2B),在3.2μM时基本完全抑制。希尔系数计算为0.6。CP-465022阻断培养的大鼠皮质细胞中红藻氨酸诱导的反应的速度相对较慢,并且取决于化合物浓度。在高浓度(即10μM)下,在2分钟内(通过添加后的第一个刺激脉冲)达到完全抑制,而在32 nM下达到稳态阻断的时间约为8分钟。在这种实验范式中,在没有化合物的情况下使用对照培养基从抑制中恢复非常缓慢。需要至少20分钟的洗涤时间才能使反应恢复到对照水平(图4B)。CP-465021是CP-465022的非活性阻转异构体(Menniti等人,2000,Welch等人,2001),对红藻氨酸诱导的电流几乎没有影响(图2C)。将皮质细胞暴露于1μM CP-465021中6-8分钟,可抑制红藻氨酸诱导的电流15±10%(n=3)。 抑制机制[1] 为了确定CP-465022诱导的AMPA受体抑制是否与激动剂竞争,我们研究了CP-465,022对一系列红藻氨酸浓度诱导的皮质神经元电流的影响。32 nM CP-465022显著抑制了10μM至10 mM红藻氨酸诱导的反应(图4A)。然而,在32 nM CP-465022存在的情况下,红藻氨酸的EC50与不存在该化合物的情况相似(不存在的EC50=145μM;存在32 nM CPP-465022的EC50=125μM)。事实上,在这些细胞中,红藻氨酸(10mM)的浓度比红藻氨酸EC50高约70倍,无法克服该化合物的抑制作用。因此,抑制作用与激动剂没有竞争性。 接下来,我们测试了CP-465,022是否需要开放通道来抑制红藻氨酸诱导的反应。如图4B所示,这是通过在没有激动剂刺激的情况下将培养的皮质神经元暴露于CP-465022来实现的。在建立红藻氨酸诱导电流的基线振幅后,将细胞暴露于对照溶液、10 nM CP-465022或100 nM CP-475022中10分钟。在这10分钟内,没有对细胞施用红藻氨酸。与对照组相比,10分钟拮抗剂暴露后第一次红藻氨酸诱导反应的振幅呈剂量依赖性降低(图4B)。暴露于10 nM和100 nM CP-465022后,第一次红藻氨酸诱导的反应分别被抑制了54±6%(n=3)和88±4%(=3)。这些抑制水平与这些浓度的化合物在反复应用红藻氨酸期间产生的抑制水平相似。因此,CP-465022的抑制作用是在没有通道激活的情况下产生的。 最后,在-60 mV或+30 mV电压钳位的皮质神经元中评估了CP-465022抑制效力的电压依赖性。在这两种条件下,100 nM CP-46502对红藻氨酸诱导反应的抑制时间过程和程度相似,在-60mV(n=3)时抑制率为73±7%,在+30 mV(n=4)时抑制度为70±5%(图4C)。这表明抑制不依赖于电压。 CP-465022对AMPA受体的特异性[1] 上述数据表明,CP-465,022是AMPA受体的强效非竞争性拮抗剂。接下来,我们研究了该化合物对AMPA受体的特异性,而不是对其他配体门控离子型受体。 在皮质和小脑颗粒神经元的原代培养中,研究了CP-465022对NMDA受体介导的反应的影响。在没有Mg2+的情况下,100μM NMDA加10μM甘氨酸诱导了皮质神经元中NMDA受体的激活(图5A)。与CP-465022在1μM下孵育10分钟对NMDA诱导的峰值电流几乎没有影响(0±0%抑制,n=3),但在NMDA应用过程中8秒测得的电流降低了26±5%。10μM的CP-465022抑制了皮质神经元中NMDA诱导的峰值电流36±4%,8秒时测量的电流70±10%(n=6)。这些抑制水平在3-4次NMDA应用(3-4分钟)内明显处于稳定状态。我们没有进一步研究CP-465022阻断峰值和持续NMDA诱导电流的差异。我们还研究了CP-465022对培养的大鼠小脑颗粒神经元中NMDA诱导电流的影响(图5B)。1μM的CP-465022在10分钟内抑制了这些细胞中的峰值NMDA电流,平均抑制率为19±11%,8秒时测量的NMDA电流为45±9%(n=4),与皮质神经元中观察到的情况相似。 |
| 体内研究 (In Vivo) |
CP-465022能有效地抑制AMPA受体介导的海马突触传递和癫痫发作的诱导。然而,在相当的剂量下,CP-465,022未能预防短暂全脑缺血后CA1神经元的损失,也未能减少暂时性大脑中动脉闭塞后的梗死体积。
结论:鉴于CP-465022对AMPA的选择性高于红藻氨酸和N-甲基-D-天冬氨酸亚型的谷氨酸受体,该化合物缺乏神经保护作用,这对缺血后AMPA受体抑制的神经保护作用提出了质疑。[2]
AMPA受体介导的体内突触传递[2] 施加于Schaeffer侧支/连合通路的刺激在对侧CA1区诱发了约3 mV的可重复群体尖峰(图1A)。这种反应是由CA1神经元上的AMPA和NMDA受体介导的,正如在海马切片制备中所确立的那样。CP-465022有效且可逆地降低了诱发群体尖峰的振幅(图1A)。当静脉注射1分钟时,这种效果呈剂量依赖性(图1B)。最大抑制量为1mg/kg,在约30分钟内逆转。皮下给药后,抑制作用持续了更长的时间(图1C)。外消旋CP-465022在15mg/kg SC时观察到最大抑制作用(相当于7.5mg/kg的拆分的S对映体);在较高剂量下,有时会观察到致命性。皮下注射喹喔啉二酮AMPA受体拮抗剂YM-90K也可逆地降低了诱发的群体尖峰的振幅。在56 mg/kg SC的最大有效剂量下(图1C),抑制程度与CP-465022相似。然而,YM-90K的抑制作用比CP-465022更快,持续时间更短。 戊四唑引发的癫痫发作[2] 对大鼠施用戊四唑(100mg/kg IP)会导致阵挛性癫痫发作的特征性综合征,随后是强直性癫痫发作,并在30分钟内致死。CP-465022在戊四唑前60分钟皮下注射,剂量依赖性地增加了戊四唑诱导的阵挛发作、强直性发作和致死率的潜伏期并降低了其发生率(图3,表)。在10mg/kg的剂量下,所有3项措施均观察到完全保护。在该剂量下,预处理时间长达4小时,观察到疗效(数据未显示)。YM-90K还增加了戊四唑诱导的癫痫发作的潜伏期和发生率,并增加了致死率,尽管其效力低于CP-465022(表)。在戊四唑(56mg/kg SC)前30分钟给予完全抑制强直性发作和致死性的YM-90K剂量,如果在戊四唑醇前2小时给予,则无效(数据未显示)。 Locomotor活动[2] CP-465022剂量依赖性地降低了水平和垂直运动活动,ED50值分别为11.9和6.6 mg/kg(表)。YM-90K产生了类似的效果,但效力较低(表)。 全身缺血[2] 10分钟的全脑缺血导致7天后海马CA1锥体神经元的大量损失。19在2个剂量水平下评估了CP-465,022预防这种神经元损失的能力。在1组中,再灌注时以5mg/kg SC给药CP-465022,4小时后以2mg/kg SC给药。第二组的给药剂量相似,但分别为10和4mg/kg的CP-465022。对照组接受了溶媒注射。7天后,短暂的全身缺血导致赋形剂治疗组81%的CA1神经元丢失(图4)。CP-465022在任一测试剂量下均未能减少观察到的神经元损失。 局灶性缺血[2] 在大鼠临时MCAO模型中也检查了CP-465022的神经保护作用。16动物接受单侧MCAO,90分钟后给予5mg/kg SC的CP-465022或赋形剂。30分钟后,取出动脉夹,再过3.5小时,动物接受2 mg/kg SC或赋形剂的第二次CP-465022给药。在开始闭塞后,体温保持在37°C 6小时,在此期间CP-465022对其他生理参数的影响很小。闭塞开始24小时后,赋形剂治疗动物的皮质梗死体积为141±46 mm3(平均值±标准差)(图4)。CP-465022导致梗死体积小幅减少(至124±49 mm3),但与赋形剂对照组没有显著差异。第二组4只动物在相同条件下接受更高剂量的CP-465022(10mg/kg SC);然而,这些动物都在化合物给药后1小时内死亡。 |
| 细胞实验 |
皮质、hNT和小脑颗粒神经元的全细胞记录[1]
CP-465022对红藻氨酸诱导的电流的抑制率是浓度依赖性的,并且在洗脱后抑制是非常缓慢可逆的。因此,使用以下方案得出效力估计。如果在拮抗剂暴露之前,连续三次红藻氨酸脉冲中对红藻氨酸的电流振幅变化不超过±5%,则选择细胞进行分析。在拮抗剂暴露后,当两个连续的激动剂脉冲产生的电流振幅变化不超过±5%时,电流振幅被认为已经稳定。对于高浓度的CP-465022(例如1μM),前两个激动剂脉冲的电流幅度达到了这一标准,而对于较低浓度的化合物,3-4个脉冲后电流稳定。然后,抑制百分比被视为拮抗剂应用前脉冲和拮抗剂应用后电流稳定后第二个脉冲的电流振幅差。抑制百分比数据符合以下方程式: 其中F是以对照分数表示的反应幅度,x是化合物浓度,IC50是半最大抑制,h是希尔系数。 |
| 动物实验 |
CP-465,022和YM-90K溶解于10% Captisol(磺丁基醚β-环糊精)中。[2]
血浆中CP-465,022的浓度[2] 在氯胺酮/赛拉嗪(70:30)麻醉下,将插管插入禁食大鼠(n=5)的颈静脉。次日,动物皮下注射10 mg/kg的消旋CP-465,022。分别在给药前和给药后0.25至6小时采集血浆样本。采用高效液相色谱-紫外检测法测定血浆中CP-465,022的浓度。 全脑缺血[2] 按照先前描述的方法18,19对大鼠进行短暂的全脑缺血。低于1%至在2%氟烷麻醉下,分离双侧颈总动脉,并轻轻结扎每条血管。电凝椎动脉,并在颈椎旁肌腹侧、气管、食管、颈外静脉和颈总动脉背侧结扎。次日,在颈动脉上放置动脉瘤夹,导致大脑供血的4条主要血管闭塞(四血管闭塞)。10至15秒内观察到意识丧失。随后收紧椎旁肌周围的结扎线,以防止侧支血流通道的开放。在整个缺血期间,体温维持在37.5°C。缺血10分钟后,检查动物是否无反应以及瞳孔是否散大。动脉瘤夹已取出。切除血管后,检查血管通畅情况,并用手术夹闭合伤口。再灌注后立即以及4小时后,通过皮下注射给予动物CP-465,022或载体。动物存活7天后,用4%中性缓冲甲醛灌注固定。计数正常和异常(死亡)CA1神经元的数量,结果以死亡神经元的百分比表示,如前所述。 局灶性缺血[2] 大鼠接受暂时性MCAO,如前所述16。首先分离并永久性闭塞右侧颈总动脉。然后通过颞下入路暴露右侧大脑中动脉,并用血管夹闭塞。闭塞90分钟后以及之后4小时,通过皮下注射给予动物CP-465,022或载体。闭塞在首次给药后,继续维持30分钟(总共闭塞2小时),然后移除夹子。手术和治疗过程中温度维持在37°C。存活22小时后,将动物断头处死,迅速取出脑组织并冷冻。将脑组织切成冠状切片(厚度20 μm),切片间隔500 μm,固定于90%乙醇中,并用苏木精-伊红染色。描绘每个切片的梗死区域,并通过将连续切片的梗死面积相加并乘以切片间的间隔厚度来计算总梗死体积。 戊四唑诱发癫痫[2] 在戊四唑(100 mg/kg,腹腔注射)给药前30或60分钟给予化合物(例如CP-465,022)。然后观察大鼠的……记录30分钟内出现阵挛性癫痫发作、强直性癫痫发作和死亡的潜伏期。数据采用Kruskal-Wallis方差分析,随后进行Mann-Whitney U检验。 自发运动活性[2] 将大鼠放入装有光电管的隔音箱内的30 cm³实验箱中。在放入实验箱前,立即向未适应环境的动物施用化合物(例如CP-465,022),并记录12小时内的水平(交叉)和垂直(后肢)运动。数据采用方差分析,随后进行Dunnett's t检验,以与对照组进行多重比较。 |
| 药代性质 (ADME/PK) |
阻转异构体可能在热作用下发生外消旋化,这一过程可能使其不适用于药物开发(至少作为单一手性实体而言)。然而,CP-465,022 在 37 °C 的人血浆中具有长达 7 天的热稳定性 [1]。
CP-465,022 的血浆浓度 [2] 图 2 显示了皮下注射 10 mg/kg 外消旋 CP-465,022 后的血浆浓度。给药后约 30 分钟内浓度达到峰值,然后缓慢下降,半衰期约为 4 小时。定性观察表明,动物在给药后 30 分钟内出现共济失调,并持续约 4 小时。 |
| 参考文献 |
|
| 其他信息 |
异常的α-氨基-3-羟基-5-甲基-4-异恶唑丙酸 (AMPA) 受体活性与癫痫发生和神经退行性疾病相关的假说,促使人们寻找AMPA受体拮抗剂作为治疗这些疾病的潜在药物。我们描述了一种新型喹唑啉-4-酮类AMPA受体拮抗剂3-(2-氯苯基)-2-[2-(6-二乙氨基甲基吡啶-2-基)乙烯基]-6-氟-3H-喹唑啉-4-酮 (CP-465,022) 的功能特性。该化合物以25 nM的IC50值抑制大鼠皮层神经元中AMPA受体介导的电流。这种抑制作用与激动剂浓度无关,且不依赖于受体的使用或电压。CP-465,022对AMPA受体的选择性高于红藻氨酸受体和N-甲基-D-天冬氨酸受体。然而,研究发现该化合物对由不同AMPA受体亚基组合构成的AMPA受体具有相同的效力。这体现在CP-465,022对表达不同AMPA受体亚基的不同类型神经元中AMPA受体介导的反应的抑制效力相当。因此,CP-465,022为研究AMPA受体在生理和病理生理过程中的作用提供了一种新的工具。[1] 总之,本研究表明CP-465,022是一种新型的非竞争性AMPA受体拮抗剂。与典型的AMPA受体拮抗剂NBQX和其他喹喔啉二酮类化合物相比,该化合物对AMPA受体的特异性高于对红藻氨酸受体的特异性。在这方面,CP-465,022 与典型的非竞争性 AMPA 受体拮抗剂 GYKI 52466 和相关的 2,3-苯二氮卓类化合物相似。事实上,我们之前报道过,CP-465,022 和相关喹唑啉酮类化合物的结合位点与 2,3-苯二氮卓类化合物的结合位点重叠(Menniti 等,2000)。然而,CP-465,022 的效力显著高于后一类化合物(例如,GYKI 53655 对各种制剂中 AMPA 受体介导反应的抑制 IC50 为 1.5–6 μM;Bleakman 等,1996)。此外,CP-465,022 具有优异的药学性质和溶解性(当配制成甲磺酸盐时,CP-465,022 在 pH 4.7 的水中溶解度超过 150 mg/ml),并且外周给药后易于进入大脑(Seymour 等,1998)。因此,CP-465,022 为研究 AMPA 受体在生理和病理生理条件下的作用提供了一种新的工具。[1]
背景和目的:基于多种化合物在实验性缺血模型中对 AMPA 受体的活性(这些化合物对其他谷氨酸受体亚型的选择性各不相同),人们假设 α-氨基-3-羟基-5-甲基-4-异恶唑丙酸 (AMPA) 受体抑制剂在脑缺血后具有神经保护作用。 CP-465,022是一种新型、高效且选择性的非竞争性AMPA受体拮抗剂。本研究旨在探讨该化合物在实验性脑缺血后减少神经元丢失的能力,以探究AMPA受体抑制的神经保护潜力。 方法:为证明CP-465,022能够进入大脑,我们研究了全身给药CP-465,022对大鼠海马AMPA受体介导的电生理反应以及化学诱导癫痫发作的影响。随后,我们研究了该化合物在大鼠全脑缺血和局灶性缺血模型中的神经保护作用,所用剂量为在电生理和癫痫模型中已证实具有最大疗效的剂量。[2] 总之,本文结果表明,仅靠AMPA受体抑制可能不足以解释喹喔啉二酮类化合物对缺血性神经元损伤的显著疗效。另一方面,CP-465,022 和 YM-90K 表现出相似的中枢神经系统抑制活性,表明 AMPA 受体抑制确实是喹喔啉二酮类化合物发挥中枢神经系统抑制活性的原因。如果 AMPA 受体抑制并非神经保护作用的必要条件,那么或许可以找到一种谷氨酸受体靶点,在降低副作用的同时提供神经保护作用。CP-465,022 为开展此类研究提供了一个重要的新工具。[2] |
| 分子式 |
C26H25CL2FN4O
|
|---|---|
| 精确质量 |
498.138
|
| CAS号 |
1785666-59-2
|
| 相关CAS号 |
CP-465022;199655-36-2;CP-465022 maleate;199656-46-7
|
| PubChem CID |
53251536
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
48.8
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
34
|
| 分子复杂度/Complexity |
727
|
| 定义原子立体中心数目 |
0
|
| SMILES |
CCN(CC)CC1=NC(=CC=C1)/C=C/C2=NC3=C(C=C(C=C3)F)C(=O)N2C4=CC=CC=C4Cl.Cl
|
| InChi Key |
YKYDGCRJPYLXHY-GVYCEHEKSA-N
|
| InChi Code |
InChI=1S/C26H24ClFN4O.ClH/c1-3-31(4-2)17-20-9-7-8-19(29-20)13-15-25-30-23-14-12-18(28)16-21(23)26(33)32(25)24-11-6-5-10-22(24)27;/h5-16H,3-4,17H2,1-2H3;1H/b15-13+;
|
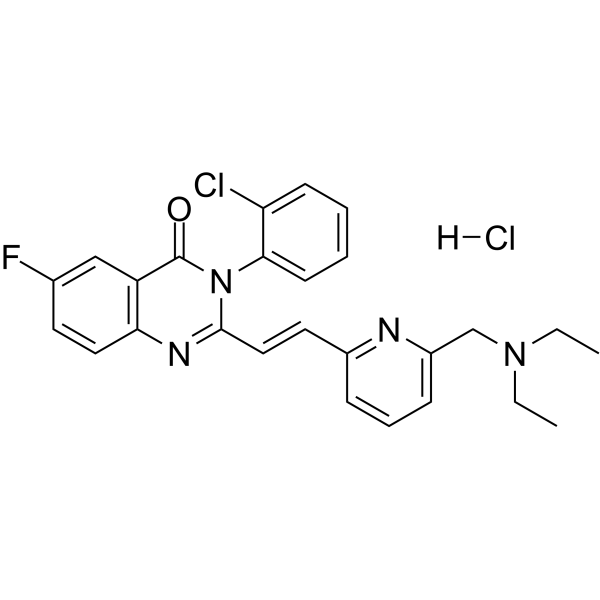
| 化学名 |
3-(2-chlorophenyl)-2-[(E)-2-[6-(diethylaminomethyl)pyridin-2-yl]ethenyl]-6-fluoroquinazolin-4-one;hydrochloride
|
| 别名 |
CP 465022 HYDROCHLORIDE; 1785666-59-2; CP465022 HCl; CP-465022 hydrochloride; 3-(2-Chlorophenyl)-2-[2-[6-[(diethylamino)methyl]-2-pyridinyl]ethenyl]-6-fluoro-4(3H)-quinazolinone hydrochloride; CP-465,022 (hydrochloride); CP 465,022; 3-(2-Chlorophenyl)-2-(2-(6-((diethylamino)methyl)pyridin-2-yl)vinyl)-6-fluoroquinazolin-4(3H)-one hydrochloride;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
