| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| Other Sizes |
|

| 靶点 |
RAS family: KRAS(G12C); c-Raf; H-Ras; NRAS rG4
|
|---|---|
| 体外研究 (In Vitro) |
RMC-7977是一种可逆的三复合RAS抑制剂,对突变型和野生型KRAS、NRAS和HRAS变体(RAS(ON)多选择性抑制剂)的活性状态具有广谱活性。临床前,RMC-777对携带各种RAS基因型的RAS-附加肿瘤表现出有效的活性,特别是对具有KRAS密码子12突变的癌症模型(KRASG12X)。[1]
RMC-7977抑制了FLT3-ITD(Molm-14,MV4-11)、KITN822K(Kasumi-1,SKNO-1)和RAS突变(NRASQ61L-OCIAML-3,HL-60,KRASG13D-NOO-1)驱动的AML细胞系的细胞增殖,IC 50值在5至33 nM之间,并抑制了MAPK途径MEK、ERK和RSK下游效应器的磷酸化。我们还评估了RMC-797在长期暴露于FLT3i后发生继发性NRASG12C或NRASQ617K突变的耐药Molm-14细胞中的活性。在低至5 nM的浓度下,RMC-7977在两种NRAS突变抗性细胞系中恢复了对吉尔特替尼的敏感性。使用胱天蛋白酶3/7测定,我们观察到RMC-7977诱导了AML细胞系的凋亡,但程度不同。我们之前已经证明,有效的MAPK信号抑制会增加对BCL2的凋亡依赖性。因此,在RMC-7977中添加venetoclax显著增强了FLT3、KIT和NRAS突变细胞系中胱天蛋白酶的激活。在细胞活力测定中,RMC-7977和venetoclax显示出高协同活性,如Bliss独立模型所评估的。[2] 鉴于患者体内肿瘤异质性与靶向治疗的临床耐药性有关,研究人员在模型中研究了RMC-7977的体外活性,这些模型再现了FLT3i治疗患者中观察到的克隆生长模式。我们将荧光标记的细胞系与FLT3-ITD(Molm-14)、FLT3-ITD和NRAS共突变(Molm-14-NRASQ61K)和仅NRAS突变(OCIAML-3)混合,用RMC-7977单独和联合(与吉尔特替尼或venetoclax)处理混合物96小时,然后通过流式细胞术评估细胞存活率。Gilteritinib和Gilteritini/venetoclax组合被选择用于携带NRAS突变的细胞的存活,但RMC-7977抑制了所有细胞群的生长。RMC-7977和吉尔特替尼的组合在含有FLT3和FLT3-NRAS共突变的混合物中具有优异的活性,但在单独携带NRAS突变的细胞中,与RMC-7997单一疗法相比没有额外的益处。令人惊讶的是,RMC-7977和venetoclax的组合在所有细胞系模型中都能有效地抑制细胞存活率,并且明显优于单独使用RMC-7997。正在进行体内研究,调查RMC-7977和RMC-7997组合在RAS突变体/FLT3i抗性患者衍生的异种移植物模型中的耐受性和活性,并将进行介绍。[2] |
| 体内研究 (In Vivo) |
RMC-7977治疗导致肿瘤消退,并且在不同的RAS-添加的临床前癌症模型中具有良好的耐受性。此外,RMC-7977抑制了KRASG12C癌症模型的生长,这些模型由于RAS途径信号的恢复而对KRAS(G12C)抑制剂具有耐药性。因此,RAS(ON)多选择性抑制剂可以靶向多种致癌和野生型RAS亚型,并有可能治疗各种RAS成瘾的癌症,这些癌症具有高度未满足的临床需求。一种相关的RAS(ON)多选择性抑制剂RMC-6236目前正在KRAS突变实体瘤患者中进行临床评估(ClinicalTrials.gov标识符:NCT05379985)。[1]
在这项研究中,研究人员评估了RMC-7977在各种PDAC模型中的治疗潜力。我们观察到,在体内耐受良好的暴露条件下,直接抑制RAS后,模型具有广泛而明显的抗肿瘤活性。药理学分析显示,肿瘤组织与正常组织对RMC-7977的反应不同。治疗后的肿瘤表现出凋亡波和持续的增殖停滞,而正常组织的增殖仅短暂下降,没有凋亡的证据。在本地KPC小鼠模型中,RMC-7977治疗导致生存期显著延长,随后治疗复发。对复发肿瘤的分析表明,Myc拷贝数增加是一种普遍的候选耐药机制,可以通过体外组合TEAD抑制来克服。这些数据共同为在PDAC环境中使用广谱RAS-GTP抑制建立了强有力的临床前理论基础,并确定了一种有前景的候选联合治疗方案来克服单一疗法的耐药性[3]。 |
| 酶活实验 |
RAS–RAF和RAS–CYPA TR-FRET[1]
如前所述,使用时间分辨荧光共振能量转移(TR-FRET)来评估野生型RAS或突变致癌RAS蛋白与BRAF的RAS结合结构域之间相互作用的破坏,并评估RAS蛋白与CYPA12之间相互反应的诱导。 CYPA结合亲和力[1] 如前所述,化合物对CYPA(Kd1)的结合亲和力通过Biacore 8K仪器上的SPR进行评估12。 RAS结合亲和力[1] 在Biacore 8K仪器上通过SPR评估化合物结合的CYPA对上述突变致癌RAS蛋白(Kd2)的结合亲和力。AviTag RAS[残基1-169]固定在链霉抗生物素蛋白传感器芯片上,并在测定缓冲液(10 mM HEPES NaOH pH 7.4,150 mM NaCl,0.005%v/v表面活性剂P20,2%v/v DMSO,25μM CYPA)中使不同浓度的化合物流过芯片。使用稳态亲和力模型或1:1结合(动力学)模型拟合SPR传感图,以评估RAS结合的解离常数(Kd)。 |
| 细胞实验 |
细胞RAS–RAF和RAS–CYPA测定[1]
将U2OS细胞或具有PPIA基因敲除的U2OS细胞以每孔500000个细胞的速度接种在6孔板中,并孵育过夜。将含有指定突变的KRAS4B或其他小GTP酶克隆到pNLF-N或pHTN质粒中,分别用N端纳米萤光素酶或HaloTag融合物表达。将全长CYPA克隆到pHTN中,将RAF1的RBD(残基51-149)克隆到pHTC中,将全长RALGDS克隆到pHTC中,将PIK3CA克隆到pNLF-N中,将SOS1的催化结构域(残基558-1049)克隆到pNL F-N中。按照制造商的方案,使用Fugene HD试剂将KRAS和效应质粒转染U2OS细胞,将小GTP酶和CYPA质粒转染U2OSPPIA-KO细胞。第二天,用胰蛋白酶收集细胞,并在含有4%FBS和1:1000稀释的NanoBRET 618 HaloTag配体的OptiMem无酚红培养基(Gibco)中的白色组织培养处理的96孔板中重新接种。对于终点浓度响应曲线,在含有4%FBS的OptiMem无酚红培养基中,将间日嗪纳米萤光素酶底物添加到1倍浓度。在Perkin-Elmer Envision平板读数器上测量纳米BRET信号之前,添加不同浓度的抑制剂并孵育1或4小时。对于动力学分析,使用去哌嗪纳米萤光素酶底物代替间日嗪,并将平板放置在预平衡至37°C和5%CO2的Cytation5多模式阅读器中。平衡1小时后,加入RMC-7977(50 nM)并测量纳米BRET信号。 PRISM分析[1] RMC-7977在博德研究所建立的931个PRISM DNA条形码细胞系中进行了筛选。简而言之,每个池中有20-25个细胞系被放置在384孔板中,并用RMC-7977以8个剂量以3倍稀释,从10µM开始处理5天。然后在TCL mRNA裂解缓冲液中裂解细胞,然后进行逆转录PCR。然后如前所述进行条形码检测和单变量和多变量分析43。数据分析见补充方法。我们分析的最新代码位于github链接:https://github.com/cmap/dockerized_mts. View More
Cell panel[1] 细胞增殖分析[1] 将细胞接种在2D的384孔或96孔组织培养处理板中,并孵育过夜。或者,将细胞接种在圆底超低附着96孔板中,以1000rpm离心10分钟以沉淀细胞,并孵育过夜或长达72小时以形成3D球体。将细胞暴露于化合物或DMSO对照(0.1%v/v)的连续稀释液中120小时。根据制造商的方案,通过CellTiter Glo 2.0试剂(2D CTG)(Promega,G9243)或3D CellTiter Glo试剂(3D CTG)测定细胞存活率。使用Perkin-Elmer Enspire的SpectraMax M5平板阅读器检测发光。将发光信号归一化为载体处理的孔(归一化信号(%)=(发光(处理)/平均发光(载体))×100%)。对于PSN1和HUPT3,原始信号被归一化为车辆控制和低信号控制化合物((样本信号-平均低控制信号)/(平均车辆信号-平均高控制信号)×100%)。对于用RMC-7977和桑格列菲林A竞争性CYPA抑制剂(3 mM)组合处理的NCI-H441和AsPC-1细胞,发光信号被标准化为仅用CYPA抑制剂处理的对照的发光信号(标准化信号(%)=(发光(处理)/平均发光(仅限CYPA抑制剂)×100%)。 细胞和上清液的生物分析[1] 1000万个细胞在37°C下暴露于1×106个细胞ml−1的悬浮液中的RMC-7977(10、100或1000 nM)1小时。通过离心将细胞制成颗粒,保留1ml上清液并在-80°C下冷冻。细胞颗粒在冷PBS中洗涤两次,在干冰和乙醇的浆液中快速冷冻之前,称量含有细胞颗粒的预先称重的试管。通过液相色谱-串联质谱(LC-MS/MS)方法测定细胞沉淀和上清液中RMC-7977的浓度。将细胞沉淀样品重新悬浮在细胞培养基中(根据需要稀释),然后作为上清液处理。用3倍体积的含有内标特非那定(2.5 ng ml−1)的乙腈淬灭上清液或再悬浮细胞(50µL)的等分试样。样品被涡旋、离心,并在配备岛津AD LC系统的Sciex 6500+三重四极杆质谱仪上进行分析。采用Waters ACQUITY UPLC BEH C4 1.7µm(2.1×50 mm)柱,梯度洗脱进行化合物分离。使用多重反应监测通过正电喷雾电离检测RMC-7977和内标(RMC-7977:m/z 865.273/833.500;特非那定:m/z 471.939/436.300)。定量下限为0.25 ng ml-1,校准范围为0.25至400 ng ml-1。使用每个细胞颗粒的质量(减去空管的质量)和已知的细胞数量计算RMC-7977的细胞内浓度,假设细胞的体积约为2000µm3,细胞的密度约为水的密度(因此,细胞体积=细胞质量);并且CYPA-KO细胞中任何超过培养基浓度的化合物都可能是膜结合的。对于每种测试的RMC-7977浓度,确定细胞颗粒中的化合物浓度与培养基中的化合物的比率。 小鼠细胞活力测定[3] 将具有KrasG12C或KrasG12D突变的PDAC小鼠细胞系(初始治疗或来源于RMC-7977治疗的终点KPC肿瘤)以2×103接种在96孔板中。24小时后,用DMSO或RMC-7977、ERK抑制剂(SCH772984)或MEK抑制剂(曲美替尼)的系列稀释液处理细胞。72小时后,根据制造商的说明,通过使用CellTiter Glo发光细胞活力测定法测量ATP水平来评估细胞活力。或者(在比较幼稚和抗性细胞系的实验中),使用钙黄绿素AM对活细胞进行荧光标记(在500 nMr下孵育20分钟),并使用SpectraMax i3X多模式检测平台进行计数。每个生物复制品都进行了三次技术复制,每个细胞系总共进行了3-4次生物复制。通过将药物处理值标准化为DMSO对照来计算生长百分比,DMSO对照设置为100%。从GraphPad Prism中的生物复制中生成了四参数药物反应曲线。绘制了每种测试稀释液的平均值±标准差。 对于RMC-7977处理过的幼稚和抗性细胞系的协同评估测试,使用了类似的方案,但进行了以下更改:细胞系接种后24小时,使用D300e数字分配器将RMC-7997、IAG933(2714434-21-4)或联合处理添加到细胞中。使用R Studio中的SynergyFinder软件包,使用Excess over Bliss方法计算每个细胞系的平均协同值。 人细胞系增殖试验[3] 作为Crown Bioscience筛选的各种组织类型的人类癌症细胞系的一部分,测试了19个PDAC细胞系对RMC-7977的敏感性。这些PDAC细胞系携带KRASG12D、KRASG12V、KRASG22C、KRASQ61H和BRAFV487_P492delinsA突变。为了测量细胞增殖的抑制作用,将细胞在甲基纤维素中培养,并用由帝肯D300e数字分配器分配的RMC-7977(最高浓度为1µM)或DMSO的连续稀释液处理三次。在使用CellTiter Glo测量ATP水平之前,将细胞孵育120小时。每个生物复制品都进行了三次技术复制,每个细胞系共进行了3-4次生物复制。通过将药物处理值标准化为DMSO对照来计算生长百分比,DMSO对照设置为100%。将标准化CTG测定读数绘制为对数摩尔抑制剂浓度的函数,并将四参数S形浓度-反应模型拟合到数据中。绘制了每种测试稀释液的平均值±标准差。 将携带野生型KRAS或KRASQ61H的PDAC细胞系以每孔500-4000个细胞的速度接种在透明平底96孔板上,并在使用D300e数字分配器添加指定浓度的RMC-7977或DMSO之前生长24小时。处理后,将细胞再孵育3-5天,之后使用Calcein AM对活细胞进行荧光标记(在500 nM下孵育20分钟),并使用SpectraMax i3X多模式检测平台进行计数。实验在第0天使用独立的培养板进行标准化。通过将药物处理值标准化为DMSO对照来计算生长百分比,DMSO对照设置为100%。将四参数S型浓度-反应模型拟合到至少三个生物重复的数据中。绘制了每种测试稀释液的平均值±标准差。 为了评估RMC-7977和IAG933的协同作用,使用了类似的方案,但有以下变化:细胞系接种后24小时,RMC-7997、IAG933(2714434-21-4),或使用D300e数字分配器(帝肯)将组合添加到细胞中。每个细胞系被视为一个单独的生物复制品(n=8)。使用R(Studio)中的SynergyFinder软件包,使用超额幸福法计算每个细胞系的平均协同值。 蛋白质印迹分析[3] 在生长培养基中,以每孔7.5×103至4×106个细胞的速度将细胞接种在6孔板或100毫米培养皿中。孵育过夜后,加入指定化合物(RMC-7977、IAG933或DMSO(0.1%v/v))并孵育指定时间点。用冰冷的PBS洗涤细胞两次,并用NP-40裂解缓冲液(赛默飞世尔,J60766)、MSD-Tris溶血缓冲液(MSD,R60TX-2)、RIPA缓冲液(50 mM Tris-HCl,pH 7.5,150 mM NaCl,1%NP-40,0.5%脱氧胆酸钠,0.1%SDS)或含有1%Triton X-100、20 mM Tris-HCl、150 mM NaCl和1 mM EDTA的裂解缓冲液裂解细胞。所有裂解缓冲液都补充了蛋白酶和磷酸酶抑制剂。在4°C下以21000g离心10分钟之前,刮取并收集裂解物。通过BCA测定定量含蛋白质的上清液,并在95°C下用LDS和还原剂变性等量的蛋白质。样品在12%或4-12%的Bis-Tris聚丙烯酰胺凝胶上分离,然后使用iBlot 2.0系统或湿转移转移转移到硝化纤维或PVDF膜上。在4°C下用第一抗体探测过夜之前,将膜在Intercept TBS缓冲液(Li-Cor,927-60001)或3-5%的牛奶中封闭。根据需要添加二抗,并在Li-Cor Odyssey成像仪上对膜进行成像。或者,将膜与HRP连接的二抗一起孵育,并使用ChemiDoc XRS+或ChemiDoc MP成像仪用Clarity或ClarityMax化学发光底物进行显影。 |
| 动物实验 |
RMC-7977 制剂[3]
体外研究中,RMC-7977 用 DMSO(Fisher Bioreagents,BP231-100)重悬,并以 10 mM 的储备液浓度使用。体内研究中,RMC-7977 的配制方法为:DMSO/PEG 400/Solutol HS15/水,体积比为 10/20/10/60。所有对照组均使用相同的溶剂配制。 体内异种移植研究[3] RMC-7977 治疗:荷瘤小鼠随机分组(每组 n = 3–10)。每日通过灌胃给予溶剂或 RMC-7977,剂量为 10 mg kg−1,持续 21–28 天。若肿瘤负荷达到人道终点,则提前终止研究。研究期间每周测量两次体重。瀑布图中绘制的是平均值±标准误。在单剂量药代动力学-药效学研究中,小鼠被随机分组(每剂量和时间点 n = 3–6)。分别以 10 mg kg⁻¹、25 mg kg⁻¹ 或 50 mg kg⁻¹ 的剂量口服单剂量 RMC-7977。在指定时间点收集组织(包括肿瘤、结肠和皮肤),并用 10% 福尔马林固定、包埋于最佳切割温度 (OCT; Sakura, 4583) 溶液中或液氮速冻,以备后续分析。全血转移至 K2EDTA 微量采血管(BD,365974)中,孵育 5 分钟后,立即用液氮速冻。 体内同种异体移植研究[3] 小鼠研究:所有小鼠同种异体移植研究以及与动物操作、护理和治疗相关的程序均符合机构动物护理和使用委员会 (IACUC) 的所有适用法规和指南。本研究使用来自杰克逊实验室的 6-8 周龄雌性 C57BL/6J(000664 品系)小鼠。同种异体移植模型的构建:为了构建皮下同种异体移植肿瘤,将 3 × 10⁵ 个 KPCY 6499c4 肿瘤细胞悬浮于 0.1 ml Matrigel:PBS (1:1) 混合液中,接种于每只小鼠的右侧腹部。当平均肿瘤体积达到 140 mm³ 时开始治疗。使用数字游标卡尺测量肿瘤的二维尺寸,并使用公式体积 = (宽度² × 长度)/2 计算肿瘤体积(单位:mm³)。每周对实验小鼠进行两次称重并测量肿瘤大小。为了构建原位同种异体移植瘤,将 5 × 10⁴ 个 KPCY 6499c4 肿瘤细胞悬浮于 20 µl PBS/Matrigel 混合物(1:1)中,通过腹腔镜切口直接植入小鼠胰腺。当平均肿瘤体积达到约 50 mm³ 时开始治疗。每周两次测量小鼠体重并通过超声监测肿瘤生长情况。 RMC-7977 治疗:将荷瘤小鼠随机分组(每组 n = 9–10),并每日通过灌胃给予载体或 RMC-7977(10 mg kg⁻¹)。对于皮下KPCY研究,生存终点定义为:肿瘤体积达到2000 mm³或小鼠出现任何临床症状,包括严重溃疡。对于原位KPCY研究,生存终点定义为:(1)小鼠出现任何临床症状,包括弓背或腹腔积液;或(2)肿瘤尺寸超出超声成像范围。研究期间每周测量两次小鼠体重。分别在末次给药后 4 小时或 24 小时收集组织,并按先前所述方法保存(参见“体内异种移植研究”)。 查看更多体内 GEMM 研究[3] KPC 小鼠药效学研究 [3] 每两周触诊一次,监测 KPC 小鼠的肿瘤形成情况。超声检测到直径为 4–7 mm 的肿瘤后,将 KPC 小鼠随机分组,分别给予载体(n = 6)或 RMC-7977(50 mg kg⁻¹;n = 11)治疗。每隔一天通过灌胃给药,持续 1 周。每日检查小鼠健康状况和体重,并每隔三天进行一次超声检查(Vevo 3100)以监测肿瘤生长。连续两次超声检查后,RMC-7977 治疗组小鼠在末次给药后 4 小时(n = 7)或 24 小时(n = 4)处死,载体对照组小鼠在末次给药后 4 至 24 小时内处死。组织收集和保存方法如前所述(参见“体内异种移植研究”)。另取一组 KPC 小鼠,单次给予 RMC-7977(n = 10)或载体(n = 3),并在给药后 4 小时或 24 小时收集组织,方法如前所述。 KPCY 小鼠药效学研究[3] 通过超声检测到 15–100 mm3 的肿瘤后,将 KPCY 小鼠纳入研究。小鼠被随机分组,分别接受载体(n = 6)或RMC-7977(25 mg kg−1;n = 8)治疗。每日通过灌胃给药,持续15天,并在第8天和第15天进行超声检查。末次给药后处死小鼠,并收集组织,按照先前描述的方法进行保存(参见“体内异种移植研究”)。 KPC小鼠生存研究[3] 在生存研究中,纳入肿瘤直径为4-7 mm(超声测量)的KPC小鼠,并隔日分别接受载体(n = 9)或RMC-7977(50 mg kg−1;n = 13)治疗。每日检查小鼠的健康状况和体重,并每三天进行一次超声检查以监测肿瘤生长情况。生存终点由总体健康标准评分确定,终点由以下标准评分达到 5 分或更高确定:濒死,立即实施安乐死;出血性腹水引起的腹胀,5 分;轻度呼吸困难,5 分;体温过低,5 分;乳糜性腹水引起的腹胀,3 分;体重下降超过入组体重的 20%,3 分;抓握测试失败,3 分;黄疸或苍白,3 分;抓握测试无力,2 分;无法与其他小鼠互动,1 分;弓背,1 分;竖毛/无法梳理毛发,1 分。 小鼠血液和肿瘤样本生物分析[1] 采用 LC-MS/MS 方法测定 RMC-7977 在全血和肿瘤中的浓度。将肿瘤组织样本用10倍体积的甲醇/15 mM PBS(1:2,v/v)匀浆。样本制备和分析在配备ACQUITY UPLC系统的Sciex 6500+三重四极杆质谱仪上进行,方法如前所述12。采用正离子电喷雾电离多反应监测(MRM)检测RMC-7977和内标维拉帕米(RMC-7977:m/z 865.4/706.4;维拉帕米:m/z 455.2/164.9)。 |
| 参考文献 | |
| 其他信息 |
RAS癌基因(统称NRAS、HRAS,尤其是KRAS)是癌症中最常见的突变基因之一,常见的驱动突变发生在12、13和611密码子。KRAS(G12C)癌蛋白的小分子抑制剂已在多种癌症患者中显示出临床疗效,并已获批用于治疗非小细胞肺癌2,3。然而,KRAS(G12C)突变仅占KRAS突变型癌症的约15%4,5,且目前尚无获批的KRAS抑制剂可用于治疗大多数携带其他常见KRAS突变的肿瘤患者。本文介绍RMC-7977,一种可逆的三复合物RAS抑制剂,对突变型和野生型KRAS、NRAS和HRAS变体的活性状态均具有广谱活性(一种RAS(ON)多选择性抑制剂)。临床前研究表明,RMC-7977 对携带多种 RAS 基因型的 RAS 依赖性肿瘤具有显著的活性,尤其对携带 KRAS 12 号密码子突变(KRASG12X)的癌症模型效果显著。RMC-7977 治疗可导致肿瘤消退,且在多种 RAS 依赖性临床前癌症模型中均表现出良好的耐受性。此外,RMC-7977 还能抑制对 KRAS(G12C) 抑制剂耐药的 KRASG12C 癌症模型的生长,这归因于其恢复了 RAS 通路信号传导。因此,RAS(ON) 多选择性抑制剂可以靶向多种致癌性和野生型 RAS 亚型,并有望治疗多种 RAS 依赖性癌症,满足其巨大的未满足临床需求。一种相关的RAS(ON)多选择性抑制剂RMC-6236目前正在KRAS突变型实体瘤患者中进行临床评估(ClinicalTrials.gov注册号:NCT05379985)[1]。FLT3抑制剂(FLT3i),例如吉瑞替尼,在急性髓系白血病(AML)中具有临床活性,但由于RAS/MAPK突变克隆的出现而导致的耐药性限制了其应用。这些突变克隆可能单独存在,也可能与FLT3突变或其他驱动基因(例如KIT突变)联合存在。RAS突变也常与IDH1/2抑制剂和BCL2抑制剂维奈托克治疗后的复发相关。重要的是,在复发时会发现多种异质性耐药克隆。此外,5-10%的新发AML患者携带致癌性RAS突变。携带 RAS 突变或其他激活 RAS/MAPK 信号通路突变的患者无法从已获临床批准的靶向治疗中获益。靶向致癌 RAS 一直以来都极具挑战性,而抑制 MAPK 通路下游效应因子(例如 MEK)在临床试验中显示出疗效有限且毒性较高。因此,有效抑制 RAS/MAPK 通路是急性髓系白血病 (AML) 治疗中亟待解决的关键问题。RMC-7977 是一种强效的口服小分子抑制剂,可同时抑制野生型和突变型 GTP 结合的 RAS 癌蛋白 (RAS MULTI),是临床候选药物 RMC-6236 的临床前研究化合物,目前正在进行临床评估 (NCT05379985)。RMC-7977 与细胞内分子伴侣环孢亲和素 A 非共价结合,形成一种对所有 RAS 亚型均具有高亲和力的新型结合界面。由此形成的三元复合物通过空间位阻阻断RAS-效应蛋白相互作用,从而抑制致癌信号的传递。我们报告了体外数据,支持RAS MULTI(ON)抑制剂在携带RAS突变的急性髓系白血病(AML)模型中的临床前应用价值,包括那些由于RAS信号过度激活而对FLT3抑制剂产生耐药性的模型。鉴于患者肿瘤异质性与靶向治疗的临床耐药性相关,我们研究了RMC-7977在能够重现FLT3抑制剂治疗患者中观察到的克隆增殖模式的模型中的体外活性。我们将荧光标记的细胞系混合,这些细胞系分别携带 FLT3-ITD 突变(Molm-14)、FLT3-ITD 和 NRAS 共突变(Molm-14 NRASQ61K)以及仅携带 NRAS 突变(OCIAML-3)。随后,我们分别用 RMC-7977 单独处理以及与吉瑞替尼或维奈托克联合处理这些混合物 96 小时,并通过流式细胞术评估细胞活力。吉瑞替尼和吉瑞替尼/维奈托克联合用药均能促进携带 NRAS 突变的细胞存活,而 RMC-7977 则抑制所有细胞群的增殖。RMC-7977 与吉瑞替尼联合用药在含有 FLT3 和 FLT3-NRAS 共突变的混合物中表现出更优的活性,但在仅携带 NRAS 突变的细胞中,其疗效并不优于 RMC-7977 单药治疗。值得注意的是,RMC-7977 与维奈托克联合用药在所有细胞系模型中均能有效抑制细胞活力,且效果显著优于 RMC-7977 单药治疗。目前正在进行体内研究,以评估 RMC-7977 及其联合用药在 RAS 突变/FLT3i 耐药的患者来源异种移植模型中的耐受性和活性,相关结果将在后续报告中公布。总而言之,我们的数据提供了临床前证据,表明利用 RAS MULTI(ON) 抑制的联合疗法能够有效抑制 RAS 突变型 AML 克隆,而 RAS 突变是 AML 中目前已获批准的靶向疗法的常见耐药机制,也是目前临床上亟待解决的重大问题。[2]
广谱 RAS 抑制剂有望使大约四分之一的 RAS 突变驱动型癌症患者获益。 RMC-7977 是一种对 KRAS、HRAS 和 NRAS 的活性 GTP 结合形式具有高度选择性的抑制剂,对突变型和野生型变体均有亲和力。超过 90% 的人类胰腺导管腺癌 (PDAC) 病例是由 KRAS 的激活突变驱动的。[3] |
| 分子式 |
C47H60N8O6S
|
|---|---|
| 分子量 |
865.094309806824
|
| 精确质量 |
864.44
|
| 元素分析 |
C, 65.25; H, 6.99; N, 12.95; O, 11.10; S, 3.71
|
| CAS号 |
2765082-12-8
|
| PubChem CID |
164726623
|
| 外观&性状 |
White to light yellow solid powder
|
| LogP |
4.7
|
| tPSA |
172Ų
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
12
|
| 可旋转键数目(RBC) |
8
|
| 重原子数目 |
62
|
| 分子复杂度/Complexity |
1620
|
| 定义原子立体中心数目 |
5
|
| SMILES |
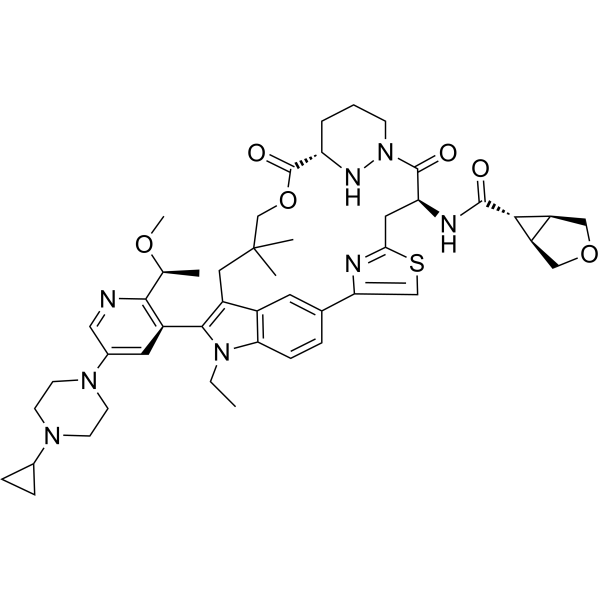
CCN1C2=C3C=C(C=C2)C4=CSC(=N4)C[C@@H](C(=O)N5CCC[C@H](N5)C(=O)OCC(CC3=C1C6=C(N=CC(=C6)N7CCN(CC7)C8CC8)[C@H](C)OC)(C)C)NC(=O)C9[C@H]1[C@@H]9COC1
|
| InChi Key |
NBLZKEHVVJSAAY-OFTZCYAMSA-N
|
| InChi Code |
InChI=1S/C47H60N8O6S/c1-6-54-39-12-9-28-18-31(39)33(43(54)32-19-30(22-48-42(32)27(2)59-5)53-16-14-52(15-17-53)29-10-11-29)21-47(3,4)26-61-46(58)36-8-7-13-55(51-36)45(57)37(20-40-49-38(28)25-62-40)50-44(56)41-34-23-60-24-35(34)41/h9,12,18-19,22,25,27,29,34-37,41,51H,6-8,10-11,13-17,20-21,23-24,26H2,1-5H3,(H,50,56)/t27-,34-,35+,36-,37+,41+/m0/s1
|
| 化学名 |
(1R,5S,6r)-N-((63S,4R,Z)-12-(5-(4-cyclopropylpiperazin-1-yl)-2-((S)-1-methoxyethyl)pyridin-3-yl)-11-ethyl-10,10-dimethyl-5,7-dioxo-61,62,63,64,65,66-hexahydro-11H-8-oxa-2(4,2)-thiazola-1(5,3)-indola-6(1,3)-pyridazinacycloundecaphane-4-yl)-3-oxabicyclo[3.1.0]hexane-6-carboxamide
|
| 别名 |
2765082-12-8; SCHEMBL25774785; RMC-7977; RMC 7977; RMC7977; EX-A7974;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.1559 mL | 5.7797 mL | 11.5595 mL | |
| 5 mM | 0.2312 mL | 1.1559 mL | 2.3119 mL | |
| 10 mM | 0.1156 mL | 0.5780 mL | 1.1559 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 GGTI-286
GGTI-286
 SOS1-IN-3
SOS1-IN-3
 KRAS G12C inhibitor 44
KRAS G12C inhibitor 44
 Rac1 Inhibitor W56
Rac1 Inhibitor W56
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
