| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
Abl1 6.95 μM (IC50); KIT 2.45 μM (IC50); PDGFR 1.39 μM (IC50)
|
|---|---|
| 体外研究 (In Vitro) |
除了PDGFRα激酶抑制剂2(化合物1)外,所有其他衍生物都失去了对ABL的活性,可能是由于伊马替尼(36)的哌嗪环上带正电的甲基化氮与Ile 789和His 790 (KIT激酶编号)的羰基之间的离子相互作用丧失。另一方面,含有不同亲电陷阱的化合物保留了对KIT和PDGFR的活性。为了确认在伊马替尼类似物和目标半胱氨酸残基的γ-硫之间形成共价加合物,用质谱(MS)分析了不同抑制剂孵育的激酶的胰蛋白酶消化。含有氯乙酰胺基团的化合物3确实导致含有目标半胱氨酸残基的片段消失,并出现与KIT和PDGFRα的肽抑制剂加合物相对应的新峰。进一步的MS/MS片段测序表明,与化合物3反应的半胱氨酸加合物的分子量(450 amu)发生了预期的变化(图4D)。其他化合物没有显示出类似的共价加合物。这可以通过亲电试剂的不太理想的定位来合理化(例如,化合物1战斗部与硫原子之间的角度不利于SN2取代;图4C)或质谱分析缺乏灵敏度。【1】
|
| 酶活实验 |
Human Kinase Data Set[1]
478种人类真核蛋白激酶的491激酶催化结构域对应的序列及其序列比对文件从Manning等人发表的数据中检索。为了本研究的目的,这种对齐已经被改进(可根据要求提供)。对于每个激酶,通过BIRD系统从蛋白质数据库下载相应的晶体结构(2010年7月)。工作流程的一部分(图3)解释了从晶体结构中提取的所有链的数据清洗,以及对构象的分类,在Leproult等人(待提交)中有详细介绍。这导致了总共1357条链,其中974条、250条和133条链分别被分类为活性构象、c -螺旋外构象和dfg -外构象。 ATP结合位点口袋检测[1] 每个链都是用PDB2PQR程序制备的,该程序可以有效地自动去除配体、溶剂和离子。这也被用来以一种与有利氢键一致的方式给所有的原子加氢。为了自动检测链内的空口袋,使用了内部的Pck程序。该程序致力于基于几何的检测和表征口袋。这使用alpha形状理论来表示蛋白质的表面,然后实现特定于某些类型口袋的算法。在这些算法中,CAST被优先选择,因为它能够检测部分埋藏的口袋,正如激酶ATP结合位点所观察到的那样。由于对构象的分类需要所有链的叠加(待提交,Leproult等),因此通过测试其是否包含一个虚拟创建的3D点,放置在铰链区域附近的ATP结合位点,来定位ATP结合位点对应的口袋。最后,只有23条链被报道没有检测到ATP结合位点口袋。对这些链的进一步检查显示,它们中的大多数具有小口袋或侧链,阻断部分ATP结合位点,导致自抑制现象。 ATP结合位点内的氨基酸选择[1] 在具有参与所检测的ATP口袋的原子的氨基酸中,那些具有至少一个参与该口袋的侧链原子的氨基酸被选择用于该工作流程的下一部分。对于那些只有主链原子参与结合位点的分子,利用DSSP程序考虑了二级结构信息。事实上,如果氨基酸位于一个灵活的结构元件上,例如一个环或一个弯,那么氨基酸被选择。唯一的特殊情况是甘氨酸氨基酸位于非柔性结构元件上。因为甘氨酸氨基酸没有侧链,所以研究了与Cα原子相连的两个氢。如果在半胱氨酸侧链突变时产生R手性的那一个参与检测到的ATP结合位点口袋,则选择甘氨酸。 稳健性氨基酸位置和对所有人类激酶的繁殖[1] 在序列比对文件中,每个先前选择的氨基酸在相应的激酶序列中突出显示。这就提供了进入氨基酸位置的途径。一旦获得了所有链的氨基酸位置,那么相对于构象类,在所有链中出现超过30%的氨基酸位置被认为是稳健位置。这一步骤避免了保留位于异常扩展的ATP结合位点的罕见位置。事实上,Pck之所以能检测到一些大的ATP结合位点,是因为ATP结合位点与其他结构域接近,或者在链中缺乏形成ATP结合位点的结构元件。接下来,使用序列比对将健壮的氨基酸位置传播到所有人类激酶。这允许推断具有侧链的氨基酸参与ATP结合位点的每一个人类激酶的每一个构象。稳健的半胱氨酸位置是本研究的重点,可以在以下网站上以3D方式可视化:http://lbgi.igbmc.fr/Kinatown. ABL1 wt、KIT wt和PDGFRα wt激酶的IC50抑制作用[1] 采用放射蛋白激酶测定法(33PanQinase活性测定法)测定ABL1 wt、KIT wt和PDGFRα wt激酶(Proqinase, Freiburg, Germany)的激酶活性。所有激酶检测均在Perkin-Elmer (Boston, MA)的96孔FlashPlates上进行,使用50 μL实验缓冲液(60 mM hepe - naoh, pH 7.5, 3 mM MgCl2, 3 mM MnCl2, 3 μM Na3VO4, 1.2 mM DTT, 50 μg mL−1 PEG2000和1 μM [γ-33P]ATP), 20 ng激酶和通用底物(用于KIT的polyGluTyr和用于ABL和PDGFRα的polyAlaGluLsTyr)和1% DMSO。试验化合物浓度范围为20 μM ~ 0.1 nM(半对数稀释)。通过将ATP溶液与测试化合物和ad预混合进行检测。 |
| 参考文献 | |
| 其他信息 |
Kinases have emerged as one of the most prolific therapeutic targets. An important criterion in the therapeutic success of inhibitors targeting the nucleotide binding pocket of kinases is the inhibitor residence time. Recently, covalent kinase inhibitors have attracted attention since they confer terminal inhibition and should thus be more effective than reversible inhibitors with transient inhibition. The most robust approach to design irreversible inhibitors is to capitalize on the nucleophilicity of a cysteine thiol group present in the target protein. Herein, we report a systematic analysis of cysteine residues present in the nucleotide binding site of kinases, which could be harnessed for irreversible inhibition, taking into consideration the different kinase conformations. We demonstrate the predictive power of this analysis with the design and validation of an irreversible inhibitor of KIT/PDGFR kinases. This is the first example of a covalent kinase inhibitor that combines a pharmacophore addressing the DFG-out conformation with a covalent trap.
|
| 分子式 |
C24H20CLN5O
|
|---|---|
| 分子量 |
429.90
|
| 精确质量 |
429.135
|
| CAS号 |
404844-11-7
|
| PubChem CID |
10181075
|
| 外观&性状 |
Solid powder
|
| 密度 |
1.3±0.1 g/cm3
|
| 熔点 |
300ºC
|
| 折射率 |
1.691
|
| LogP |
4.04
|
| tPSA |
79.8
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
31
|
| 分子复杂度/Complexity |
570
|
| 定义原子立体中心数目 |
0
|
| SMILES |
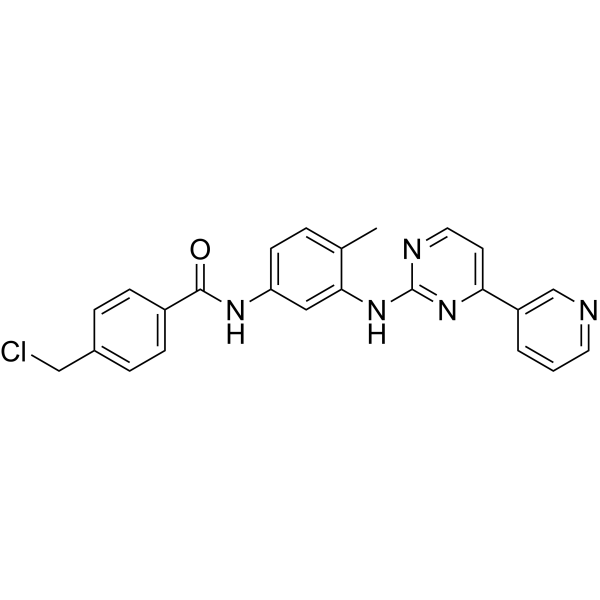
O=C(NC1=CC=C(C)C(NC2=NC=CC(C3=CC=CN=C3)=N2)=C1)C4=CC=C(CCl)C=C4
|
| InChi Key |
WUGWDFMSJPJUEZ-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C24H20ClN5O/c1-16-4-9-20(28-23(31)18-7-5-17(14-25)6-8-18)13-22(16)30-24-27-12-10-21(29-24)19-3-2-11-26-15-19/h2-13,15H,14H2,1H3,(H,28,31)(H,27,29,30)
|
| 化学名 |
4-(chloromethyl)-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide
|
| 别名 |
404844-11-7; 4-(Chloromethyl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)benzamide; 4-Chloromethyl-N-[4-methyl-3-[[4-(pyridin-3-yl)pyrimidin-2-yl]amino]phenyl]benzamide; N-[4-Methyl-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-phenyl]-4-chloromethyl Benzamide; 4-(chloromethyl)-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide; PDGFR; A kinase inhibitor 2; CHEMBL1765715;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : 25 mg/mL (58.15 mM; with sonication (<60°C))
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3261 mL | 11.6306 mL | 23.2612 mL | |
| 5 mM | 0.4652 mL | 2.3261 mL | 4.6522 mL | |
| 10 mM | 0.2326 mL | 1.1631 mL | 2.3261 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 cSRC/BCR-ABL-IN-1
cSRC/BCR-ABL-IN-1
 BCR-ABL-IN-11
BCR-ABL-IN-11
 cSRC/BCR-ABL1-IN-1
cSRC/BCR-ABL1-IN-1
 BCR-ABL-IN-10
BCR-ABL-IN-10
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
