| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| 500mg |
|
||
| Other Sizes |
|

| 靶点 |
Varenicline dihydrochloride targets α4β2 neuronal nicotinic acetylcholine receptor (nAChR) as a partial agonist, with a Ki value of 0.14 nM (radioligand binding assay) [2]
Varenicline dihydrochloride targets α7 nAChR as a full agonist, with an EC₅₀ value of 1.2 μM (ion channel activation assay) [2] |
|---|---|
| 体外研究 (In Vitro) |
Vannicline diHClide(1 μM,24 小时)可抑制 RAW 264.7 巨噬细胞 LPS 诱导的细胞因子分泌(IL-1β、IL-6 和 TNFα)和细胞增殖率 [1]。当暴露于 250 nM vannicline diHClide 时,从男性和女性器官捐献者分离的人肾上腺嗜铬细胞在没有 ACh 刺激的情况下表现出动作电位 (Aps) 刺激 [3]。通过降低 VE-钙粘蛋白表达,vannicline diHClide(100 μM,4 小时)刺激 HUVEC 迁移 [4]。
在LPS刺激的RAW 264.7巨噬细胞中:Varenicline dihydrochloride(1–10 μM)呈剂量依赖性抑制炎症细胞因子产生,TNF-α分泌减少35–68%,IL-6分泌减少40–72%(ELISA检测);同时抑制NF-κB p65核转位和p38 MAPK磷酸化(Western blot验证) [1] - 在人脐静脉内皮细胞(HUVECs)中:Varenicline dihydrochloride(0.1–5 μM)促进细胞迁移2.1–3.8倍(划痕实验),下调血管内皮钙粘蛋白(VE-cadherin)表达45–70%(Western blot),并通过α7 nAChR激活ERK1/2和p38 MAPK磷酸化 [4] - 在人肾上腺嗜铬细胞中:治疗浓度(0.1–1 μM)的Varenicline dihydrochloride与尼古丁(1 μM)共同存在时,动作电位发放频率增加2.5倍,动作电位时程延长(膜片钳电生理记录) [3] - 在α4β2 nAChR上:Varenicline dihydrochloride(0.01–1 μM)在表达人α4β2受体的非洲爪蟾卵母细胞中诱导部分离子通道激活(为尼古丁最大反应的40%) [2] - 在α7 nAChR上:Varenicline dihydrochloride(0.1–10 μM)在表达人α7受体的卵母细胞中诱导完全离子通道激活(与尼古丁等效) [2] - 该化合物在浓度高达20 μM时,对RAW 264.7细胞、HUVECs或肾上腺嗜铬细胞无明显细胞毒性 [1][3][4] |
| 体内研究 (In Vivo) |
在尼古丁(0.5 mg/kg 皮下注射)之前 10 分钟给药时,万尼克林 disalk(0.01-1 mg/kg 皮下注射,3 天)可抑制尼古丁条件性位置偏好 (CPP) [5]。由vannicline diHClide(皮下注射,2.5 mg/kg,3天)引起的位置厌恶依赖于α5 nAChR,但不依赖于β2 nAChR [5]。皮下注射vannicline diHClide(0.1和0.5 mg/kg,3天)以剂量相关的方式逆转与尼古丁戒断相关的躯体症状和痛觉过敏以及戒断引起的厌恶[5]。
在尼古丁致敏的C57BL/6小鼠模型中:口服Varenicline dihydrochloride(1、3 mg/kg,每日1次,连续7天),呈剂量依赖性降低尼古丁诱导的条件性位置偏爱(CPP)评分,分别降低32%和58% [5] - 在小鼠尼古丁戒断模型中:Varenicline dihydrochloride(3 mg/kg口服)减少戒断相关行为(跳跃次数减少65%),缓解尼古丁诱导的痛觉过敏,热板实验中热退缩潜伏期增加42% [5] - 该化合物单独给药时,未在小鼠中诱导条件性位置偏爱或厌恶效应 [5] |
| 酶活实验 |
α4β2 nAChR结合实验:将表达人α4β2 nAChR的细胞膜与[³H]-尼古丁及系列稀释的Varenicline dihydrochloride在25°C孵育2小时,通过过滤分离结合态放射性配体,检测放射性强度计算Ki值 [2]
- α7 nAChR功能实验:向非洲爪蟾卵母细胞注射编码人α7 nAChR的cRNA,孵育2–3天后加入Varenicline dihydrochloride,采用双电极电压钳记录离子电流,确定EC₅₀和激动剂效能 [2] |
| 细胞实验 |
细胞增殖测定 [1]
细胞类型: RAW 264.7 小鼠巨噬细胞(用 4 μg/mL LPS 处理 24 小时) 测试浓度: 1 μM 孵育时间:0-48小时 实验结果:LPS诱导的细胞增殖率减弱。 蛋白质印迹分析[4] 细胞类型: HUVEC 测试浓度: 1、10、100 μM 孵育持续时间:24 小时或 30 分钟 实验结果:VE-钙粘蛋白表达减少,ERK1/2、p38 和 JNK 信号传导激活。 巨噬细胞炎症实验(RAW 264.7):细胞接种于24孔板,用Varenicline dihydrochloride(1–10 μM)预处理1小时后,加入1 μg/mL LPS刺激24小时。收集培养上清液通过ELISA定量TNF-α/IL-6;细胞裂解液用于Western blot检测NF-κB p65和p38 MAPK [1] - 内皮细胞迁移实验(HUVECs):细胞培养至汇合后,用移液管尖端划痕,加入Varenicline dihydrochloride(0.1–5 μM),在0和24小时通过图像分析软件测量迁移距离;Western blot检测VE-cadherin和MAPK磷酸化水平 [4] - 电生理实验(人肾上腺嗜铬细胞):分离并培养细胞,用Varenicline dihydrochloride(0.1–1 μM)联合尼古丁(1 μM)处理,采用全细胞膜片钳技术记录动作电位 [3] |
| 动物实验 |
动物/疾病模型: ICR雄性小鼠[5]
剂量: 0.01-1 mg/kg,3天 给药途径: 皮下注射 实验结果: 以剂量依赖的方式抑制尼古丁条件性位置偏好(CPP)。 尼古丁CPP小鼠模型:雄性C57BL/6小鼠(20-25 g)每天一次腹腔注射尼古丁(0.5 mg/kg)致敏,持续7天。盐酸伐尼克兰在每次尼古丁注射前30分钟口服(1、3 mg/kg)。在第 8 天,使用双室装置测试条件性位置偏好 (CPP) [5] - 尼古丁戒断和痛觉过敏模型:将小鼠植入尼古丁缓释片 (7.2 mg) 14 天以诱导尼古丁依赖。取出缓释片以诱发戒断反应,并每日一次口服给予盐酸伐尼克兰 (3 mg/kg),连续 3 天。记录 30 分钟内的戒断行为(跳跃);通过热板试验评估热痛觉过敏 [5] - 药物制剂:将盐酸伐尼克兰溶解于生理盐水中进行口服给药 [5] |
| 药代性质 (ADME/PK) |
吸收、分布和排泄
伐尼克兰代谢极少,92%以原形经尿液排出。伐尼克兰主要通过肾小球滤过和肾小管主动分泌(可能通过有机阳离子转运蛋白OCT2)进行肾脏清除。 口服伐尼克兰后,血浆浓度通常在3-4小时内达到峰值。多次口服伐尼克兰后,4天内即可达到稳态血药浓度。在推荐剂量范围内,单次或重复给药后,伐尼克兰的药代动力学呈线性关系。质量平衡研究表明,口服伐尼克兰后吸收几乎完全,全身生物利用度约为90%。食物或给药时间不影响伐尼克兰的口服生物利用度。伐尼克兰的血浆蛋白结合率较低(≤20%),且与年龄和肾功能无关。 伐尼克兰主要以原形药物经尿液排泄。药物的肾脏排泄主要通过肾小球滤过和肾小管主动分泌进行。 伐尼克兰在动物体内会分泌到乳汁中。尚不清楚伐尼克兰是否会分泌到人乳中。 代谢/代谢物 代谢有限(<10%)。大部分活性成分经肾脏排泄(81%)。少量伐尼克兰会发生葡萄糖醛酸化、氧化、N-甲酰化以及与己糖结合等代谢。 伐尼克兰代谢极少,92%以原形经尿液排出。 伐尼克兰代谢极少,92%以原形经尿液排出,不到10%以代谢物形式排出。尿液中的少量代谢物包括伐尼克兰N-氨甲酰葡萄糖醛酸苷和羟基伐尼克兰。在循环系统中,伐尼克兰占药物相关物质的91%。循环中的少量代谢物包括伐尼克兰N-氨甲酰葡萄糖醛酸苷和N-葡萄糖基伐尼克兰。 生物半衰期 伐尼克兰的消除半衰期约为24小时。 伐尼克兰的消除半衰期约为24小时。 |
| 毒性/毒理 (Toxicokinetics/TK) |
肝毒性
伐尼克兰治疗期间血清酶升高发生率并未高于安慰剂组,但关于这些异常的信息有限,且有报道称偶有无症状ALT升高导致停药的病例。在数千例患者参与的上市前关键性注册试验中,伐尼克兰未引起黄疸或肝炎。上市后,有少数病例报告显示,在开始服用伐尼克兰4周内出现血清酶升高但无黄疸,但这些病例大多发生于其他肝损伤病因(酒精性肝病、丙型肝炎)的患者。该肝损伤具有自限性,且与免疫过敏或自身免疫特征无关。在冰岛,已报告一例伐尼克兰肝毒性病例(病例1),据估计,自该药上市以来,冰岛已有约20,000人接受过该药治疗。 可能性评分:C(可能是临床上明显的肝损伤的罕见原因)。 妊娠和哺乳期影响 ◉ 哺乳期用药概述 伐尼克兰是一种尼古丁部分激动剂,口服用于辅助戒烟,鼻喷剂用于治疗干眼症。一位研究人员指出,根据动物实验中尼古丁的数据,伐尼克兰可能会干扰婴儿正常的肺部发育,因此不建议哺乳期妇女使用。由于目前尚无关于哺乳期使用伐尼克兰的信息,因此建议使用其他药物,尤其是在哺乳新生儿或早产儿时。然而,鼻喷剂对母亲的药物暴露量仅为口服药物的 7.5% 左右,因此鼻喷剂对婴儿的影响要小得多。如果母亲在服用伐尼克兰期间选择母乳喂养,应密切监测婴儿是否出现癫痫发作和过度呕吐。 ◉ 对母乳喂养婴儿的影响 截至修订日期,未找到相关的已发表信息。 ◉ 对泌乳和母乳的影响 截至修订日期,未找到相关的已发表信息。 蛋白质结合 低于 20%。 体外毒性:在 RAW 264.7 巨噬细胞、HUVEC 和人肾上腺嗜铬细胞中,CC₅₀ > 20 μM [1][3][4] - 体内急性毒性:口服剂量高达 50 mg/kg 的伐尼克兰二盐酸盐治疗小鼠,未观察到死亡或明显的行为异常(嗜睡、共济失调)[5] -血浆蛋白结合率:10-20%(人血浆,超滤法)[2] |
| 参考文献 |
[1]. Elif Baris, et al. Varenicline Prevents LPS-Induced Inflammatory Response via Nicotinic Acetylcholine Receptors in RAW 264.7 Macrophages. Front Mol Biosci. 2021 Oct 12;8:721533.
[2]. Mihalak KB, et al. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors.Mol Pharmacol. 2006 Sep;70(3):801-5. Epub 2006 Jun 9. [3]. Jin H, et al. Therapeutic concentrations of varenicline in the presence of nicotine increase action potential firing in human adrenal chromaffin cells. J Neurochem. 2017 Jan;140(1):37-52. [4]. Mitsuhisa Koga, et al. Varenicline promotes endothelial cell migration by lowering vascular endothelial-cadherin levels via the activated α7 nicotinic acetylcholine receptor-mitogen activated protein kinase axis. Toxicology. 2017 Sep 1;390:1-9. [5]. Bagdas D, et al. New insights on the effects of varenicline on nicotine reward, withdrawal and hyperalgesia in mice.Neuropharmacology. 2018 Aug;138:72-79. |
| 其他信息 |
伐尼克兰是一种用于治疗烟瘾的处方药。它是首个获批的尼古丁受体部分激动剂。具体而言,伐尼克兰是尼古丁乙酰胆碱受体α4/β2亚型的部分激动剂。此外,它还作用于α3/β4受体,对α3β2和α6亚型受体的作用较弱。它对α7受体表现出完全激动作用。2015年3月9日,美国食品药品监督管理局(FDA)发出警告,辉瑞公司生产的戒烟药物畅沛(Chantix)中的伐尼克兰成分与癫痫发作有关,并且一些服用该药期间饮酒的患者可能会出现攻击性行为或昏厥。辉瑞公司正在对该药物进行一项额外的安全性研究,预计结果将于2015年底公布。FDA表示,至少在试验结果公布之前,将维持黑框警告。
伐尼克兰是尼古丁乙酰胆碱受体的部分激动剂,用于辅助戒烟。伐尼克兰治疗期间血清酶升高的发生率较低,并且自获批并广泛使用以来,仅有极少数临床表现明显的轻度肝损伤病例。 伐尼克兰是尼古丁乙酰胆碱受体(nAChR)α4β2亚型的部分激动剂。尼古丁刺激位于伏隔核突触前末端的中枢α4β2 nAChR,导致神经递质多巴胺的释放,这可能与愉悦感有关;尼古丁成瘾是一种与多巴胺奖赏系统相关的生理依赖。伐尼克兰作为一种乙酰胆碱受体(AChR)部分激动剂,可以减轻戒断尼古丁引起的渴求和戒断症状,但其本身不具有成瘾性。 伐尼克兰是一种苯并氮杂卓衍生物,作为α4β2尼古丁受体部分激动剂发挥作用。它用于戒烟。 另见:盐酸伐尼克兰(注释已移至此处)。 药物适应症 用于辅助戒烟。伐尼克兰鼻喷剂适用于干眼症的症状治疗。 FDA标签 作用机制 伐尼克兰是一种α4β2神经元尼古丁乙酰胆碱受体部分激动剂。该药物对该受体亚类具有高度选择性,远高于其他尼古丁受体(α3β4 >500 倍,α7 >3500 倍,α1βγδ >20000 倍)或非尼古丁受体和转运蛋白(>2000 倍)。该药物竞争性抑制尼古丁与 α4β2 受体的结合和激活。该药物在该位点具有轻微的激动活性,但远低于尼古丁;据推测,这种激活作用可以缓解戒断症状。 伐尼克兰是一种选择性 α4β2 尼古丁乙酰胆碱受体部分激动剂。该药物与位于大脑中的α4β2尼古丁乙酰胆碱受体具有高亲和力和选择性,并能刺激受体介导的活性,但其作用强度远低于尼古丁;1,6这种低水平的受体刺激以及随后中脑边缘多巴胺的适度、持续释放被认为可以减轻与戒烟相关的渴求和戒断症状。伐尼克兰还能阻断尼古丁激活α4β2受体的能力,从而阻止尼古丁诱导的中脑边缘多巴胺能系统的刺激,进而降低吸烟的强化和奖赏效应。 ……α4β2神经元尼古丁乙酰胆碱受体(nAChR)部分激动剂作为治疗烟草成瘾的新型疗法的理论基础和设计。此类药物有望发挥双重作用:一方面,通过充分刺激α4β2-nAChR介导的多巴胺释放来降低戒烟时的渴求;另一方面,通过抑制吸烟时的尼古丁强化作用来发挥作用。在临床前模型中,可以筛选出具有双重激动剂和拮抗剂活性的强效且选择性的α4β2-nAChR部分激动剂。α4β2-nAChR部分激动剂伐尼克兰的临床疗效证明了该方法的有效性,其戒烟率显著高于其他疗法,为戒烟药物治疗提供了一种新的选择。 ……伐尼克兰已被证实是α4β2受体的部分激动剂,并且在平衡结合试验中,它对α4β2受体具有高度选择性。本研究检测了伐尼克兰在非洲爪蟾卵母细胞中表达的多种大鼠神经元尼古丁受体上的功能活性,并采用双电极电压钳技术进行测定。伐尼克兰是α4β2受体的强效部分激动剂,EC50为2.3±0.3 μM,效能(相对于乙酰胆碱)为13.4±0.4%。伐尼克兰对α3β4受体的效力较低,但效能较高,EC50为55±8 μM,效能为75±6%。伐尼克兰似乎也是α3β2和α6受体的弱部分激动剂,效能低于10%。值得注意的是,伐尼克兰是α7受体的强效完全激动剂,其EC50值为18±6 μM,效力为93±7%(相对于乙酰胆碱)。因此,尽管伐尼克兰在某些异源神经元尼古丁受体上是部分激动剂,但它却是同源α7受体的完全激动剂。伐尼克兰作为戒烟辅助药物的作用机制可能涉及上述某些作用的组合。 盐酸伐尼克兰是一种合成的神经元尼古丁乙酰胆碱受体调节剂,临床上用于戒烟[2][5] - 其在巨噬细胞中的抗炎机制涉及α7 nAChR的激活,抑制NF-κB和MAPK信号通路,从而减少促炎细胞因子的产生[1] - 该化合物通过α7 nAChR介导的VE-钙黏蛋白下调和MAPK(ERK1/2、p38)的激活促进内皮细胞迁移[4] - 在戒烟方面,它作为α4β2 nAChR的部分激动剂(降低尼古丁渴求)和α7 nAChR的完全激动剂(调节奖赏通路)发挥作用,同时阻断尼古丁与α4β2结合[2][5] - 在尼古丁存在的情况下,它增强肾上腺嗜铬细胞的动作电位发放,可能调节儿茶酚胺的释放[3] |
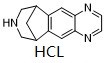
| 分子式 |
C13H15CL2N3
|
|---|---|
| 分子量 |
284.1843
|
| 精确质量 |
247.088
|
| 元素分析 |
C, 54.94; H, 5.32; Cl, 24.95; N, 14.79
|
| CAS号 |
866823-63-4
|
| 相关CAS号 |
Varenicline;249296-44-4;Varenicline-d4 hydrochloride;Varenicline-d4 dihydrochloride
|
| PubChem CID |
45263226
|
| 外观&性状 |
Brown to dark brown solid powder
|
| LogP |
2.934
|
| tPSA |
37.81
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
0
|
| 重原子数目 |
18
|
| 分子复杂度/Complexity |
254
|
| 定义原子立体中心数目 |
0
|
| SMILES |
Cl.N1C2C(=CC3C4CC(CNC4)C=3C=2)N=CC=1
|
| 别名 |
Varenicline dihydrochloride; HSDB7591; HSDB-7591; HSDB 7591; CP 526555; CP-526555; CP526555;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 请将本产品存放在密封且受保护的环境中(例如氮气保护),避免吸湿/受潮和光照。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO : ~62.5 mg/mL (~219.93 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.5189 mL | 17.5945 mL | 35.1890 mL | |
| 5 mM | 0.7038 mL | 3.5189 mL | 7.0378 mL | |
| 10 mM | 0.3519 mL | 1.7594 mL | 3.5189 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Botswana Smoking and Abstinence Reinforcement Trial
CTID: NCT05694637
Phase: Phase 4 Status: Enrolling by invitation
Date: 2024-11-13
 Nanofin-d2
Nanofin-d2
 Nanofin-d2 hydrochloride
Nanofin-d2 hydrochloride
 Catestatin (rat)
Catestatin (rat)
 Nanofin-d5
Nanofin-d5
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
