| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 50mg |
|
| 靶点 |
ENaC; uTPA; polycystin-2 (TRPP2)
|
|---|---|
| 体外研究 (In Vitro) |
阿米洛利阻断 δβγ 通道的 IC50 为 2.6 μM (58, 71, 75, 134, 148)。阿米洛利对 δβγ ENaC 通道的 Ki 比 αβγ 通道大 26 倍(αβγ ENaC 为 0.1 μM)。与 αβγ 通道相比,阿米洛利对 δβγ ENaC 的阻断更具电压依赖性。与 αβη 和 δβγ 通道的 Ki 值相反,δαβγ 通道的阿米洛利 Ki 值在 -120 和 +80 mV 下分别为 920 和 13.7 μM [1]。阿米洛利是一种相对选择性的上皮钠通道 (ENaC) 抑制剂。其 IC50(即导致离子通道 50% 堵塞所需的剂量)介于 0.1 至 0.5 μM 之间。在外部 [Na+] 含量低的情况下,IC50 低至 3 μM,在 [Na+] 高的情况下,IC50 高达 1 mM,阿米洛利是 Na+/H+ 交换器 (NHE) 的相对较弱的抑制剂。阿米洛利的 IC50 为 1 mM,使其成为 Na+/Ca2+ 交换器 (NCX) 较弱的抑制剂。已知阿米洛利 (1 μM) 和亚微摩尔剂量的苯扎米尔 (30 nM) 会抑制 ENaC。这意味着通过阻止 ENaC 蛋白发挥作用,它们可以阻止对灌注压升高的肌源性血管收缩反应。在血管平滑肌细胞 (VSMC) 中,阿米洛利完全阻止 Na+ 流入,其剂量已知对 ENaC 具有相对选择性 (1.5 μM) [2]。
|
| 体内研究 (In Vivo) |
在饮用盐水、易发生中风的自发性高血压大鼠 (SHRSP) 中,与对照组相比,阿米洛利改善了大脑和肾脏的组织学评分,逆转了胶原蛋白沉积的最初增加,并通过皮下注射(1 毫克/公斤/天)防止了该组织的进一步增加)[2]。阿米洛利还拮抗或阻断醛固酮在这些细胞以及患有盐依赖性高血压的动物的心血管和肾组织中的作用。
|
| 细胞实验 |
MR和ENaC已在成纤维细胞、VSMC和内皮细胞内的mRNA和蛋白质水平上得到证实。Kornel等人发现,用生理剂量醛固酮(5 nmol/L)治疗7至10天的兔子主动脉VSMC的Na+内流显著增加。此外,在已知对ENaC相对特异的剂量(1.5μmol/L)下,阿米洛利几乎完全抑制了Na+内流,但NHE特异性阿米洛利类似物乙基异丙基阿米洛利或NCX特异性二氯苯甲酰胺都没有。在另一项研究中,用醛固酮(2mg/天)治疗兔子4周。随后,从主动脉中分离VSMC,并使用ENaC特异性[3H]阿米洛利结合对Na+通道进行定量。醛固酮治疗的动物VSMC Na+通道数量增加了一倍,表明VSMC内醛固酮和ENaC之间可能存在关系。[2]
|
| 动物实验 |
Campbell et al examined myocardial fibrosis in a high-salt/aldosterone state. Uninephrectomized Sprague-Dawley rats were placed on 1% NaCl drinking solution and given one of the following for 8 weeks: 1) aldosterone 0.75 μg/h subcutaneously; 2) amiloride 1 mg/kg/day subcutaneously; 3) aldosterone + amiloride subcutaneously; or 4) vehicle. Aldosterone increased BP significantly, an effect that was attenuated by amiloride. Microscopic scarring, a reparative fibrosis indicative of myocyte loss, was apparent in animals treated with aldosterone. However, when amiloride was given simultaneously with aldosterone, the microscopic scarring of both the left and right ventricles was completely prevented. The finding of scarring to the nonhypertensive right ventricle suggests that the benefits of amiloride were not from BP-lowering effects alone. The authors postulated that myocyte necrosis in hyperaldosteronism is likely a result of enhanced potassium excretion that can be prevented by amiloride. Although this remains a possibility, measurements of urinary Na+/K+handling and serum K+ were not performed. Furthermore, subsequent studies suggest other possible mechanisms of benefit (see later here).[2]
|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Readily absorbed following oral administration. Amiloride HCl is not metabolized by the liver but is excreted unchanged by the kidneys. About 50 percent of a 20 mg dose of amiloride HCl is excreted in the urine and 40 percent in the stool within 72 hours. Metabolism / Metabolites Amiloride is not metabolized by the liver but is excreted unchanged by the kidneys. Biological Half-Life Plasma half-life varies from 6 to 9 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Hepatotoxicity
Amiloride therapy has not been associated with serum aminotransferase elevations. Idiosyncratic, clinically apparent liver injury from amiloride is rare, but several instances have been reported as isolated case reports in which the combination of amiloride with hydrochlorothiazide was used (Case 1). The numbers of cases have been too few to characterize the clinical features, but the latency to onset was ranged from 2 to 12 months and the pattern of injury either hepatocellular or mixed. Immunoallergic features and autoantibodies have not been associated with the liver injury from amiloride. Likelihood score: D (possible rare cause of liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the use of amiloride during breastfeeding, so an alternate drug may be preferred. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Intense diuresis can suppress lactation; however, it is unlikely that amiloride alone is sufficiently potent to cause this effect. |
| 参考文献 |
|
| 其他信息 |
Amiloride is a member of the class of pyrazines resulting from the formal monoacylation of guanidine with the carboxy group of 3,5-diamino-6-chloropyrazine-2-carboxylic acid. It has a role as a sodium channel blocker and a diuretic. It is a member of pyrazines, an organochlorine compound, an aromatic amine and a member of guanidines. It is a conjugate base of an amiloride(1+).
A pyrazine compound inhibiting sodium reabsorption through sodium channels in renal epithelial cells. This inhibition creates a negative potential in the luminal membranes of principal cells, located in the distal convoluted tubule and collecting duct. Negative potential reduces secretion of potassium and hydrogen ions. Amiloride is used in conjunction with diuretics to spare potassium loss. (From Gilman et al., Goodman and Gilman's The Pharmacological Basis of Therapeutics, 9th ed, p705) Amiloride is a Potassium-sparing Diuretic. The physiologic effect of amiloride is by means of Decreased Renal K+ Excretion, and Increased Diuresis. Amiloride is a potassium-sparing diuretic used in the therapy of edema often in combination with thiazide diuretics. Amiloride has been linked to rare cases of clinically apparent drug induced liver disease. Amiloride is a synthetic pyrazine derivative with antikaliuretic and diuretic properties. Amiloride inhibits sodium channels located in the distal tubules and collecting ducts of the kidney, thereby preventing the absorption of sodium and increasing its excretion along with water, to produce naturesis. In response to the hypernatremic conditions in the kidney, the plasma membrane becomes hyperpolarized and electrochemical forces are reduced, which then prevents the excretion of potassium and hydrogen into the lumen. A pyrazine compound inhibiting SODIUM reabsorption through SODIUM CHANNELS in renal EPITHELIAL CELLS. This inhibition creates a negative potential in the luminal membranes of principal cells, located in the distal convoluted tubule and collecting duct. Negative potential reduces secretion of potassium and hydrogen ions. Amiloride is used in conjunction with DIURETICS to spare POTASSIUM loss. (From Gilman et al., Goodman and Gilman's The Pharmacological Basis of Therapeutics, 9th ed, p705) Drug Indication For use as adjunctive treatment with thiazide diuretics or other kaliuretic-diuretic agents in congestive heart failure or hypertension. FDA Label Mechanism of Action Amiloride works by inhibiting sodium reabsorption in the distal convoluted tubules and collecting ducts in the kidneys by binding to the amiloride-sensitive sodium channels. This promotes the loss of sodium and water from the body, but without depleting potassium. Amiloride exerts its potassium sparing effect through the inhibition of sodium reabsorption at the distal convoluted tubule, cortical collecting tubule and collecting duct; this decreases the net negative potential of the tubular lumen and reduces both potassium and hydrogen secretion and their subsequent excretion. Amiloride is not an aldosterone antagonist and its effects are seen even in the absence of aldosterone. Pharmacodynamics Amiloride, an antikaliuretic-diuretic agent, is a pyrazine-carbonyl-guanidine that is unrelated chemically to other known antikaliuretic or diuretic agents. It is an antihypertensive, potassium-sparing diuretic that was first approved for use in 1967 and helps to treat hypertension and congestive heart failure. The drug is often used in conjunction with thiazide or loop diuretics. Due to its potassium-sparing capacities, hyperkalemia (high blood potassium levels) are occasionally observed in patients taking amiloride. The risk is high in concurrent use of ACE inhibitors or spironolactone. Patients are also advised not to use potassium-containing salt replacements. |
| 分子式 |
C6H8CLN7O
|
|---|---|
| 分子量 |
229.63
|
| 精确质量 |
229.048
|
| 元素分析 |
C, 31.38; H, 3.51; Cl, 15.44; N, 42.70; O, 6.97
|
| CAS号 |
2609-46-3
|
| 相关CAS号 |
Amiloride hydrochloride;2016-88-8;Amiloride hydrochloride dihydrate;17440-83-4
|
| PubChem CID |
16231
|
| 外观&性状 |
Typically exists as Off-white to light yellow solids at room temperature
|
| 密度 |
2.11g/cm3
|
| 沸点 |
628.1ºC at 760 mmHg
|
| 熔点 |
240.5-241.5
240 °C |
| 闪点 |
333.7ºC
|
| 蒸汽压 |
1.08E-15mmHg at 25°C
|
| 折射率 |
1.884
|
| LogP |
1.271
|
| tPSA |
156.79
|
| 氢键供体(HBD)数目 |
4
|
| 氢键受体(HBA)数目 |
5
|
| 可旋转键数目(RBC) |
1
|
| 重原子数目 |
15
|
| 分子复杂度/Complexity |
279
|
| 定义原子立体中心数目 |
0
|
| SMILES |
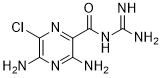
O=C(C1=NC(Cl)=C(N)N=C1N)NC(N)=N
|
| InChi Key |
XSDQTOBWRPYKKA-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C6H8ClN7O/c7-2-4(9)13-3(8)1(12-2)5(15)14-6(10)11/h(H4,8,9,13)(H4,10,11,14,15)
|
| 化学名 |
3,5-diamino-6-chloro-N-(diaminomethylidene)pyrazine-2-carboxamide
|
| 别名 |
MK-870; MK 870; Amipramidin; Guanamprazin; Guanamprazine; Amipramizid; Midamor; Amilorida; MK870
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.3548 mL | 21.7742 mL | 43.5483 mL | |
| 5 mM | 0.8710 mL | 4.3548 mL | 8.7097 mL | |
| 10 mM | 0.4355 mL | 2.1774 mL | 4.3548 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
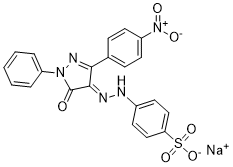 PHPS1 Sodium
PHPS1 Sodium
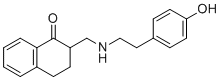 BE 2254
BE 2254
 环胞啶硫酸盐
环胞啶硫酸盐
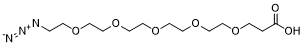 Azido-PEG5-acid
Azido-PEG5-acid
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
