| 靶点 |
VEGFR2 (IC50 = 0.2 nM); VEGFR3 (IC50 = 0.7 nM); c-Kit (IC50 = 14.8 nM); c-Kit (IC50 = 14.8 nM); c-Kit (IC50 = 14.8 nM)
|
|---|---|
| 体外研究 (In Vitro) |
体外活性:安罗替尼(原名 AL3818)是一种新型有效的多激酶抑制剂,可抑制 VEGFR2/3、FGFR1-4、PDGFRα/β、c-Kit 和 Ret。安罗替尼作为受体酪氨酸激酶 (RTK) 抑制剂,具有潜在的抗肿瘤和抗血管生成活性。安罗替尼在体外显着减少 AN3CA 细胞数量,其特点是突变 FGFR2 蛋白的高表达。每日口服安罗替尼(5 mg/kg)使 55% 的接受治疗的动物出现完全缓解,并且在 29 次治疗后,AN3CA 肿瘤的肿瘤体积和肿瘤重量分别减少了 94% 和 96%。天治疗周期。尽管卡铂和紫杉醇未能改变肿瘤生长,但与单独使用安罗替尼治疗相比,与安罗替尼的组合似乎并未表现出更好的效果。激酶检测:安罗替尼(原名 AL3818)是一种新型有效的多激酶抑制剂,可抑制 VEGFR2/3、FGFR1-4、PDGFRα/β、c-Kit 和 Ret。细胞测定:AL3818 在体外显着减少 AN3CA 细胞数量,其特点是突变 FGFR2 蛋白的高表达。每日口服 AL3818 (5 mg/kg) 可使 55% 的接受治疗动物出现完全缓解,并且在 29 次治疗后,AN3CA 肿瘤的肿瘤体积减小,肿瘤重量分别减少 94% 和 96%。天治疗周期。尽管卡铂和紫杉醇未能改变肿瘤生长,但与单独使用 AL3818 治疗相比,与 AL3818 的组合似乎并未表现出更好的效果。
|
| 体内研究 (In Vivo) |
每日口服安罗替尼(5 mg/kg)使 55% 的接受治疗的动物出现完全缓解,并且在 29 次治疗后,AN3CA 肿瘤的肿瘤体积和肿瘤重量分别减少了 94% 和 96%。天治疗周期。尽管卡铂和紫杉醇未能改变肿瘤生长,但与单独使用安罗替尼治疗相比,与安罗替尼的组合似乎并未表现出更好的效果。
|
| 酶活实验 |
安罗替尼(原名 AL3818)是一种新型强效多激酶抑制剂,可阻断 Ret、FGFR1-4、PDGFRα/β、c-Kit 和 VEGFR2/3。
如前所述,使用ELISA测定安洛替尼对酪氨酸激酶的抑制活性。22 ATP与酪氨酸激酶的反应在反应缓冲液(50 mmol/L HEPES pH 7.4,50 mmol/L MgCl2,0.5 mmol/L MnCl2,0.2 mmol/L Na3VO4,1 mmol/L DTT)中启动,并在37°C下在预涂有20μg/mL Poly(Glu,Tyr)4:1的96孔板中孵育1小时。将平板与PY99抗体一起孵育,然后与HRP偶联的抗小鼠IgG一起孵育。与邻苯二胺溶液反应后,加入2N H2SO4终止反应,使用Synergy H4 Hybrid阅读器在490 nm处测量吸光度[3]。 |
| 细胞实验 |
在体外,AL3818 显着降低了 AN3CA 细胞的数量,这是通过突变 FGFR2 蛋白的高表达来识别的。经过 29 天的治疗周期后,每天口服 AL3818(5 mg/kg),55% 的治疗动物出现完全缓解,AN3CA 肿瘤的肿瘤体积和肿瘤重量分别减少了 94% 和 96%。分别。与单独使用 AL3818 治疗相比,卡铂和紫杉醇的组合似乎没有更大的效果,尽管它们无法改变肿瘤的生长。
|
| 动物实验 |
human colon cancer SW620 xenograft model(Balb/cA-nude mice, 5-6 weeks old)
0.75, 1.5, 3 and 6 mg/kg oral Female nude mice (Balb/cA‐nude, 5‐6 weeks old), purchased from Shanghai Laboratory Animal Center (Chinese Academy of Sciences, Shanghai, China), were housed in sterile cages under laminar airflow hoods in a specific pathogen‐free room with a 12‐hour light/12‐hour dark schedule, and fed autoclaved chow and water ad libitum. Human tumor xenografts were established by s.c. inoculating cells into the left axilla of nude mice. When tumor volumes reached 100‐200 mm3, mice were divided randomly into control and treatment groups. Control groups were given vehicle alone, and treatment groups received oral anlotinib or sunitinib daily. Tumor volume was calculated as (length × width2)/2. Tumor growth inhibition was calculated from the start of treatment by comparing changes in tumor volumes for control and treatment groups.[1] Rat studies[3] Rats were randomly assigned to four groups (five male and five female rats per group) to receive a single oral dose of anlotinib at 1.5, 3, or 6 mg/kg (via gavage) or a single intravenous dose at 1.5 mg/kg (from the tail vein). Serial blood samples (around 0.25 mL; before and 5, 15, and 30 min and 1, 2, 4, 6, 8, 11, and 24 h after dosing) were collected in heparinized tubes from the orbital sinuses of rats under isoflurane anesthesia and centrifuged at 1300×g for 10 min to yield plasma fractions. Rats under isoflurane anesthesia were killed by bleeding from the abdominal aorta at 1, 4, 8, and 24 h (three male and three female rats per time point) after a single oral dose of anlotinib at 3 mg/kg. [3] Tumor-bearing mouse studies[3] Female tumor-bearing mice were randomly assigned to three groups (20 mice per group) to receive a single oral dose of anlotinib at 0.75, 1.5, or 3 mg/kg (via gavage). Mice under isoflurane anesthesia were killed by bleeding from the orbital sinus at 2, 4, 8, and 24 h (five mice per time point) after dosing. Dog study[3] Dogs were randomly assigned to four groups (three male and three female dogs per group) to receive a single oral dose of anlotinib at 0.5, 1, or 2 mg/kg (via gavage) or a single intravenous dose at 0.5 mg/kg (from left forelimb vein). |
| 药代性质 (ADME/PK) |
Plasma pharmacokinetics of anlotinib in rats and dogs [3] Figure 1 shows the change in mean plasma concentration of anlotinib over time after a single dose in rats and dogs; Table 1 summarizes the plasma pharmacokinetic parameters of anlotinib. After oral administration, in the test dose range of 1.5–6 mg/kg, the systemic exposure level of anlotinib in female rats, namely the maximum plasma concentration (Cmax) and the area under the plasma concentration-time curve (AUC0-24 h) over 24 hours, was often higher than that in male rats; while in the dose range of 0.5–2 mg/kg, there was no significant sex difference in plasma Cmax and AUC0-24 h of anlotinib in dogs. In rats and dogs, plasma Cmax and AUC0-24 h both increased proportionally with increasing anlotinib dose (Table 2). Anlotinib has a high binding rate in the plasma of rats, dogs and humans, with plasma free fractions (fu) of 2.9%, 4.0% and 7.3%, respectively. These fu values were independent of the total plasma concentration of anlotinib, indicating that the total concentration of anlotinib can well reflect changes in its free concentration in the plasma of the corresponding species. As shown in Table 3, the binding affinity of anlotinib to α1-acid glycoprotein is similar to that of another tyrosine kinase inhibitor, imatinib. However, anlotinib has a significantly higher affinity for albumin than imatinib. Unlike imatinib, whose nKα1-acid glycoprotein/nK albumin ratio is 92.0, anlotinib's ratio is 0.9 (<7.7), indicating that the plasma albumin concentration (600 μmol/L) is much higher than the α1-acid glycoprotein concentration (20 μmol/L), and this concentration difference cannot be compensated for by circulating anlotinib in the human body. Notably, anlotinib has a significantly higher affinity for plasma lipoproteins, especially low-density lipoprotein and very low-density lipoprotein, than for albumin and α1-acid glycoprotein. Following oral administration, anlotinib was rapidly absorbed from the gastrointestinal tract in both rats and dogs (Figure 1). Oral bioavailability (F) was generally higher in dogs than in rats. The terminal half-life (t1/2) after intravenous administration of anlotinib in rats and dogs was comparable to the corresponding t1/2 after oral administration. The mean t1/2 value was longer in dogs than in rats. This difference in t1/2 appears to be primarily attributed to differences in total plasma clearance (CLtot, p) between species. The mean steady-state apparent volume of distribution (VSS) of anlotinib in rats was 40 times the total body fluid volume in rats, while the VSS in dogs was 12 times the total body fluid volume in dogs, indicating that the compound is widely distributed in various body fluids and tissues. In rats receiving intravenous anlotinib, only small amounts of the unmetabolized compound were excreted in urine, bile, and feces (Table 1), indicating that metabolism is the primary pathway for anlotinib clearance.
Intestinal Absorption-Related Characteristics of Anlotinib [3] Intestinal absorption of a drug is a combination of its solubility in gastrointestinal fluids, membrane permeability, and substrate specificity to the intestinal epithelial cell efflux system. Anlotinib’s water solubility is pH-dependent, being >1 g/mL at pH 1.7 (stomach), 114 μg/mL at pH 4.6 (duodenum), and 0.89 μg/mL at pH 6.5 (jejunum and ileum). Anlotinib’s solubility at pH 1.7 and 4.6 is higher than the minimum solubility required for adequate intestinal absorption at a dose of 6 mg/kg, but its solubility at pH 6.5 is lower than that minimum solubility. The minimum solubility was derived from Lipinski's bar chart, which depicts the minimum solubility of low, medium, and high permeability compounds at doses of 0.1, 1, and 10 mg/kg. Anlotinib exhibited good membrane permeability on the monolayer membrane of Caco-2 cells expressing MDR1, MRP2, and BCRP, with a mean apparent permeability coefficient (Papp) of 3.5 × 10⁻⁶ cm/s. The mean efflux ratio (EfR) of this compound was 0.91 ± 0.22, indicating that its transcellular transport was not affected by Caco-2 apical efflux transporters (Supplementary Figure S1). The physicochemical properties of anlotinib (predicted using ACD/Percepta, Toronto, Ontario, Canada), such as molecular weight (407 Da; ideal <500 Da), hydrogen bonding ability (HBA+HBD, 6+3; <12), topological polar surface area (TPSA, 82.4 Ų; <140 Ų), and molecular flexibility (NROTB, 6; <10), all support its good membrane permeability. The LogD value was -0.89 at pH 1.7, 2.10 at pH 4.6, and 2.38 at pH 6.5 (optimal range 0–5). Tissue distribution of anlotinib in rats and tumor-bearing mice [3] After oral administration of anlotinib, the exposure levels in various tissues (measured using relevant tissue homogenate samples) in rats and tumor-bearing mice were significantly higher than the relevant systemic exposure levels (Figure 2). In rats, the lungs had the highest exposure level, which was 197 times higher than the systemic exposure level. At the same time, the liver, kidneys and heart of rats also had high exposure levels, which were 49 times, 54 times and 32 times higher than the systemic exposure level, respectively. Anlotinib can penetrate rat brain tissue, and its brain homogenate AUC0-24h is comparable to the relevant plasma concentration. In tumor-bearing mice, the exposure level of the compound in tumor tissue increased with increasing dose, which was 13 times higher than the systemic exposure level. Metabolism of Anlotinib[3] Since the parent compound is not mainly excreted via the liver, gallbladder or kidneys, the metabolites of anlotinib in rat and canine samples were detected and identified. The results showed that 12 anlotinib metabolites (M1-M12) were detected in the plasma, bile, urine and fecal samples of rats after administration (Table 4). Among them, 8 metabolites, namely M2, M4, M5, M6, M8, M9, M10 and M11, were present in plasma. Except for M7 and M11, which were only present in rat bile samples, all of these metabolites were present in both rat bile and urine samples. Five anlotinib metabolites were detected in canine plasma samples: M4, M8, M9, M10, and M11. The metabolic pathway of anlotinib was proposed after characterization of these metabolites using liquid chromatography/mass spectrometry (Figure 3). The main metabolic pathway of anlotinib in rats is likely hydroxylation to generate M10 and M11, and dealkylation to generate M8. Metabolites M10 and M11 are the two main anlotinib metabolites in rat plasma, while M8 is further glucuronidated to generate M6, which is the main anlotinib metabolite in both plasma and bile. To further characterize these metabolic pathways, we conducted in vitro metabolic studies of anlotinib. The results showed that multiple human cytochrome P450 enzymes, namely CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5, could mediate the oxidation of anlotinib to M10, M11, and M8 (Figure 4A). Among them, CYP3A4 and CYP3A5 had the strongest metabolic capacity. As shown in Figure 4B, under the same conditions, after incubating anlotinib with NADPH-enhanced rat liver microsomes, canine liver microsomes, and human liver microsomes, metabolites M10, M11, and M8 were also detected in the samples. The total amount of metabolites in liver microsomes varied among different species, indicating that the oxidation rate was highest in rat liver microsomes, followed by canine liver microsomes, and lastly in human liver microsomes. In vitro inhibitory activity of anlotinib against drug-metabolizing enzymes and transporters[3] As shown in Table 5, anlotinib showed significant inhibitory activity against CYP3A4 and CYP2C9 in vitro, with IC50 values <1 μmol/L; moderate inhibitory activity against CYP2C19, CYP2C8, UGT1A1, UGT1A4, UGT1A9 and UGT2B15, with IC50 values of 1–10 μmol/L. This tyrosine kinase inhibitor showed low inhibitory activity against human CYP2B6, CYP2D6, UGT1A6, UGT2B7, OATP1B1, OAT3, OCT2, MDR1 and BCRP in vitro, with IC50 values greater than 10 μmol/L. No significant inhibitory activity of anlotinib against human CYP1A2, CYP3A5, OATP1B3, OAT1 and MRP1 was found. Anlotinib is not an in vitro substrate for OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MDR1 and BCRP (Supplementary Table S1). |
| 毒性/毒理 (Toxicokinetics/TK) |
Under the 4/0 dosing regimen, no dose-limiting toxicities (DLTs) were observed in the first four patients starting at a dose of 5 mg/day. However, at a dose of 10 mg/day, one of the first three patients treated developed grade 3 hypertension. Subsequently, another patient was enrolled who also developed grade 3 hypertension. Therefore, dose escalation was discontinued. Meanwhile, pharmacokinetic (PK) studies showed that anlotinib continued to accumulate significantly in patients receiving continuous dosing (data not shown). Based on the pharmacokinetic characteristics of anlotinib and the two DLTs observed at a dose of 10 mg/day, we changed the dosing regimen from the 4/0 regimen to the 2/1 regimen. [2] Under the 2/1 regimen, since none of the three patients developed DLTs at the initial dose of 10 mg/day, the dose was escalated to 16 mg/day. Two of the three patients in the 16 mg dose group developed DLTs (one with grade 3 fatigue and one with grade 3 hypertension). Therefore, the maximum tolerated dose (MTD) was exceeded, and the efficacy of the next lower dose of 12 mg/day was further evaluated by enrolling more patients. None of the initial three patients experienced grade 3/4 adverse events. Based on this, a 12 mg/day dose was selected for an extension study. [2] A total of 21 patients received a 12 mg/day dose in a 2/1 dosing regimen. All patients experienced adverse events of any cause during the first two cycles. All hematologic toxicities were mild. As shown in Table 2, the most common non-hematologic adverse events included hypothyroidism, elevated triglycerides, elevated total cholesterol, elevated ALT, diarrhea, and proteinuria. Two patients (10%) experienced grade 3 adverse events during the first two cycles (one patient experienced elevated triglycerides, and one patient experienced elevated lipase). A total of six patients (29%) experienced grade 3/4 adverse events during all treatment cycles. The most common (>5%) non-hematologic grade 3 adverse events were hypertension, elevated triglycerides, hand-foot skin reaction, and elevated lipase.
|
| 参考文献 | |
| 其他信息 |
Anlotinib has been investigated for the treatment of non-small cell lung cancer and metastatic colorectal cancer. Caterquentinib is a receptor tyrosine kinase (RTK) inhibitor with potential antitumor and anti-angiogenic activities. After administration, catequentinib targets multiple RTKs, including vascular endothelial growth factor receptor type 2 (VEGFR2) and type 3 (VEGFR3). This drug may inhibit angiogenesis and prevent tumor cell growth.
Drug Indications Treatment of Ewing's sarcoma, treatment of soft tissue sarcoma Catequentinib hydrochloride is the hydrochloride form of catequentinib, a receptor tyrosine kinase (RTK) inhibitor with potential antitumor and anti-angiogenic activities. After administration, catequentinib targets multiple RTKs, including vascular endothelial growth factor receptor type 2 (VEGFR2) and type 3 (VEGFR3). This drug may inhibit angiogenesis and prevent tumor cell growth. Blocking tumor angiogenesis by inhibiting vascular endothelial growth factor receptor 2 (VEGFR2) has been established as a cancer treatment strategy. However, due to low selectivity, most small-molecule inhibitors of VEGFR2 tyrosine kinases exhibit unexpected adverse reactions and limited anticancer efficacy. In this study, we elucidate in detail the pharmacological properties of the highly selective VEGFR2 inhibitor anlotinib in preclinical models. Anlotinib occupies the ATP-binding pocket of VEGFR2 tyrosine kinases and exhibits high selectivity and inhibitory potency (IC50 < 1 nmol/L) for VEGFR2 compared to other tyrosine kinases. Consistent with this activity, anlotinib inhibited VEGF-induced signaling and cell proliferation in HUVEC cells with a picomolar IC50 value. However, in vitro experiments, anlotinib required micromolar concentrations to directly inhibit tumor cell proliferation. Anlotinib significantly inhibited HUVEC cell migration and tubular formation; it also inhibited microvascular growth in rat aortic explants in vitro and reduced vascular density in tumor tissues in vivo. Compared with the known tyrosine kinase inhibitor sunitinib, once-daily oral anlotinib showed a broader and stronger antitumor efficacy in vivo and induced tumor regression in nude mice in some models. Overall, these results suggest that anlotinib is a well-tolerated, orally effective VEGFR2 inhibitor that targets angiogenesis in tumor growth and supports continued clinical evaluation of anlotinib for a variety of malignancies. [1] Background: Anlotinib is a novel multi-target tyrosine kinase inhibitor designed to primarily inhibit VEGFR2/3, FGFR1-4, PDGFR α/β, c-Kit, and Ret. We aimed to evaluate the safety, pharmacokinetics, and antitumor activity of anlotinib in patients with advanced refractory solid tumors. Methods: Patients with solid tumors were given once-daily oral anlotinib (5-16 mg) in two dosing regimens: (1) 4 weeks of continuous treatment (4/0) or (2) 2 weeks of treatment followed by 1 week off (2/1). Pharmacokinetic sampling was performed on all patients. An expanded cohort study enrolled 21 patients to evaluate the recommended dose and dosing regimen. Preliminary tumor response was also assessed. Results: Under the 4/0 dosing regimen, the dose-limiting toxicity (DLT) was grade 3 hypertension at a dose of 10 mg. Under the 2/1 dosing regimen, the DLT was grade 3 hypertension and grade 3 fatigue at a dose of 16 mg. Pharmacokinetic evaluation indicated that anlotinib has a long elimination half-life and significant drug accumulation after multiple oral doses. Therefore, the 2/1 dosing regimen was chosen, with 12 mg once daily as the maximum tolerated dose for the expanded study. Of the 21 patients (with colon adenocarcinoma, non-small cell lung cancer, clear cell renal cell carcinoma, medullary thyroid carcinoma, and soft tissue sarcoma), 20 were evaluable for the antitumor activity of anlotinib: 3 achieved partial remission, 14 had stable disease (12 of whom experienced tumor burden reduction), and 3 experienced disease progression. Major serious adverse reactions included hypertension, elevated triglycerides, hand-foot skin reaction, and elevated lipase. Conclusion: Anlotinib, administered once daily at a dose of 12 mg in a 2/1 dosing regimen, showed manageable toxicity, a longer circulation time, and broad-spectrum antitumor potential, warranting further investigation. [2] Anlotinib is a novel oral tyrosine kinase inhibitor; this study aimed to characterize its pharmacokinetics and in vivo distribution. Anlotinib was evaluated in rats, tumor-bearing mice, and dogs, and its pharmacokinetics, in vivo distribution, and drug interaction potential were assessed in vitro. Samples were analyzed by liquid chromatography/mass spectrometry. Anlotinib has good membrane permeability and rapid absorption, with oral bioavailability of 28%-58% in rats and 41%-77% in dogs. The terminal half-life of anlotinib in dogs (22.8±11.0 h) was longer than that in rats (5.1±1.6 h). This difference appears to be primarily related to the interspecies variation in total plasma clearance (rat: 5.35 ± 1.31 L·h⁻¹·kg⁻¹; dog: 0.40 ± 0.06 L·h⁻¹/kg⁻¹). Cytochrome P450-mediated metabolism is likely the main clearance pathway. Human CYP3A exhibits the strongest metabolic capacity, while other human P450s have a smaller effect. Anlotinib showed a large apparent volume of distribution in both rats (27.6 ± 3.1 L/kg) and dogs (6.6 ± 2.5 L/kg), and high plasma binding rates in rat (97%), dog (96%), and human (93%) plasmas. In human plasma, anlotinib primarily binds to albumin and lipoproteins, rather than α1-acid glycoproteins or γ-globulins. Anlotinib concentrations in various rat tissue homogenates and tumor-bearing mouse tissue homogenates were significantly higher than the corresponding plasma concentrations. Anlotinib has limited in vitro inhibitory effects on various human P450 enzymes, UDP-glucuronyl transferases and transporters, but it does have inhibitory effects on CYP3A4 and CYP2C9 (in vitro half-maximal inhibitory concentration <1 μmol/L). According to previously reported human pharmacokinetic data, the drug interaction indices of anlotinib with CYP3A4 and CYP2C9 are 0.16 and 0.02, respectively, indicating that anlotinib is unlikely to interact with these enzymes. The pharmacokinetic characteristics of anlotinib are similar to those of other tyrosine kinase inhibitors, except for the terminal half-life, interactions with drug-metabolizing enzymes and transporters, and plasma protein binding. [3] |
| 分子式 |
C23H22FN3O3
|
|
|---|---|---|
| 分子量 |
407.45
|
|
| 精确质量 |
407.16
|
|
| 元素分析 |
C, 67.80; H, 5.44; F, 4.66; N, 10.31; O, 11.78
|
|
| CAS号 |
1058156-90-3
|
|
| 相关CAS号 |
1058156-90-3;1360460-82-7 (HCl);
|
|
| PubChem CID |
25017411
|
|
| 外观&性状 |
Solid powder
|
|
| 密度 |
1.3±0.1 g/cm3
|
|
| 沸点 |
584.4±50.0 °C at 760 mmHg
|
|
| 闪点 |
307.2±30.1 °C
|
|
| 蒸汽压 |
0.0±1.6 mmHg at 25°C
|
|
| 折射率 |
1.678
|
|
| LogP |
3.7
|
|
| tPSA |
82.4Ų
|
|
| 氢键供体(HBD)数目 |
2
|
|
| 氢键受体(HBA)数目 |
6
|
|
| 可旋转键数目(RBC) |
6
|
|
| 重原子数目 |
30
|
|
| 分子复杂度/Complexity |
606
|
|
| 定义原子立体中心数目 |
0
|
|
| SMILES |
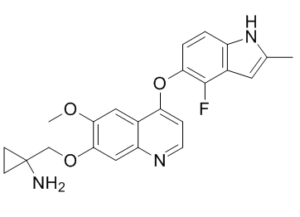
CC1=CC2=C(N1)C=CC(=C2F)OC3=C4C=C(C(=CC4=NC=C3)OCC5(CC5)N)OC
|
|
| InChi Key |
KSMZEXLVHXZPEF-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C23H22FN3O3/c1-13-9-15-16(27-13)3-4-19(22(15)24)30-18-5-8-26-17-11-21(20(28-2)10-14(17)18)29-12-23(25)6-7-23/h3-5,8-11,27H,6-7,12,25H2,1-2H3
|
|
| 化学名 |
1-[[4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxyquinolin-7-yl]oxymethyl]cyclopropan-1-amine
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4543 mL | 12.2714 mL | 24.5429 mL | |
| 5 mM | 0.4909 mL | 2.4543 mL | 4.9086 mL | |
| 10 mM | 0.2454 mL | 1.2271 mL | 2.4543 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
| NCT Number | Recruitment | interventions | Conditions | Sponsor/Collaborators | Start Date | Phases |
| NCT03890068 | Recruiting | Drug: Anlotinib Hydrochloride | Soft Tissue Sarcoma | Sun Yat-sen University | August 5, 2020 | Phase 2 |
| NCT05602415 | Recruiting | Drug: Anlotinib Procedure: Surgery |
Anlotinib Radiotherapy |
Ruijin Hospital | November 2022 | Phase 2 |
| NCT05883085 | Recruiting | Drug: Anlotinib hydrochloride | Pheochromocytoma Paraganglioma |
Peking Union Medical College Hospital |
May 1, 2022 | Phase 2 |
| NCT05218629 | Recruiting | Drug: Anlotinib, PD-1 inhibitor | Overall Survival | Qingdao Central Hospital | January 1, 2022 | Phase 2 |
| NCT05866510 | Recruiting | Drug: Utidelone and anlotinib | Esophageal Cancer | Peking University | May 15, 2023 | Phase 2 |
The lung metastasis changes in patients of alveolar soft tissue sarcoma with lung metastasis during treatment.J Hematol Oncol.2016 Oct 4;9(1):105. |
Duration of treatment and tumor size changes of 20 patients who received 12mg QD at the 2/1 schedule. J Hematol Oncol.2016 Oct 4;9(1):105. |
Plasma concentrations of anlotinib over time after a single oral dose of anlotinib capsules at 5 (green line), 10 (purple line), 12 (blue line), or 16mg anlotinib/person (red line) in male (solid circles) and female cancer patients (open circles) (a).bCorrelation of dose with plasma AUC0–120h.cCorrelation of dose with plasmaCmax.dCorrelation of dose witht1/2.ePlasma concentrations of anlotinib (24h after daily dosing) over time during multiple oral doses of anlotinib capsules at 12mg anlotinib/person/day in female cancer patients.fPlasma concentrations of anlotinib (24h after daily dosing) over time during multiple oral doses of anlotinib capsules at 12mg anlotinib/person/day in male cancer patients.J Hematol Oncol.2016 Oct 4;9(1):105. |
 Vasigrobart
Vasigrobart
 PDGFRα kinase-IN-2
PDGFRα kinase-IN-2
 PDGFRalpha V561D Recombinant Human Active Protein Kinase
PDGFRalpha V561D Recombinant Human Active Protein Kinase
 PDGFRalpha T674I Recombinant Human Active Protein Kinase
PDGFRalpha T674I Recombinant Human Active Protein Kinase
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 COA
COA



 463611831
463611831
