| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|
描述:西曲瑞克二酯(SB-75)是一种新型、高效的合成促性腺激素释放激素(GnRH)受体拮抗剂,IC50值为1.21 nM。西曲瑞克醋酸酯是一种十肽,具有用于不孕症治疗的潜力。在包括卵巢癌在内的多种人类恶性肿瘤中,已证实GnRH(GnRH-I,LHRH)及其受体作为细胞增殖自分泌调节系统的一部分而表达。 GnRH及其超激动剂类似物可呈时间和剂量依赖性地抑制人卵巢癌细胞系的增殖。
| 靶点 |
GnRH-I receptor (GnRHR) [1][2]
|
|---|---|
| 体内研究 (In Vivo) |
在裸鼠体内的人类卵巢癌OV-1063异种移植模型中,长期使用西曲瑞克(Cetrorelix)治疗可显著抑制肿瘤生长,而GnRH-I激动剂曲普瑞林则无此作用[1]。
在20例接受卵巢刺激的体外受精(IVF)患者中,从月经周期第7天至HCG注射前,皮下注射西曲瑞克(Cetrorelix),剂量分别为3 mg/天(n=15)或1 mg/天(n=5),可有效抑制内源性LH峰值,且无患者出现LH峰值提前出现的情况。平均每位患者获得8.1个卵母细胞,受精率为61.5%,并成功妊娠3例[2]。 在Balb/c小鼠中,皮下注射西曲瑞克(0.5 mg/kg/天,持续16天)可显著减少环磷酰胺诱导的卵巢卵泡破坏。在环磷酰胺 50 mg/kg 剂量下,Cetrorelix 预处理仅导致原始卵泡损失 14%,而对照组为 53% (P<0.001);在 75 mg/kg 剂量下,损失为 35%,而对照组为 54% (P<0.004)。相对保护率分别为 1.83 和 1.4 [3]。 |
| 动物实验 |
雌性Balb/c小鼠(8-9周龄,体重15-25 g)从第1天至第15天每日皮下注射0.5 mg/kg的西曲瑞克(Cetrorelix)。第9天,腹腔注射单剂量环磷酰胺(50或75 mg/kg)。对照组注射生理盐水。第16天,处死小鼠,取出双侧卵巢,用4%多聚甲醛固定,石蜡包埋,连续切片(厚度5 μm),苏木精-伊红染色,每隔10张切片计数原始卵泡(单层鳞状前颗粒细胞,无卵泡膜层,细胞核清晰可见);每只小鼠的原始卵泡总数乘以10计算得出[3]。
|
| 药代性质 (ADME/PK) |
吸收、分布和排泄
皮下注射后吸收迅速。健康女性受试者皮下给药后的平均绝对生物利用度为 85%。男性和女性皮下注射 10 mg 西曲罗利后,尿液中均检测到未代谢的西曲罗利。剂量:1.16 L/kg 剂量:1.28 ml/min·kg [健康成年女性单次皮下注射 3 mg]。男性和女性皮下注射 10 mg 西曲罗利后,尿液中均检测到未代谢的西曲罗利。24 小时后,在胆汁样本中检测到西曲罗利以及痕量的 (1-9)、(1-7)、(1-6) 和 (1-4) 肽。 2-4%的剂量以原形西曲瑞利经尿液排出,5-10%的剂量以西曲瑞利及其四种代谢物经胆汁排出。因此,24小时内仅有7-14%的总剂量以原形西曲瑞利及其代谢物的形式从尿液和胆汁中回收。由于胆汁和尿液的收集时间较短,剩余剂量可能无法回收。单次静脉注射3 mg西曲瑞利后,其分布容积约为1 L/kg。体外人血浆蛋白结合率为86%。在接受控制性卵巢刺激的患者中,取卵当日卵泡液和血浆中的西曲瑞利浓度相似。皮下注射0.25 mg和3 mg西曲瑞克后,在取卵和胚胎移植当日,血浆中西曲瑞克的浓度低于或处于定量下限。西曲瑞克皮下注射后吸收迅速,约1-2小时达到血浆峰浓度。健康女性受试者皮下注射西曲瑞克后的平均绝对生物利用度为85%。药代动力学研究主要在大鼠和犬中进行。无论性别或物种,皮下注射部位的吸收均迅速且完全。剂量-血浆AUC呈线性关系。西曲瑞克分布迅速。其主要靶器官为肾脏、肝脏、小肠以及含有促黄体生成素释放激素(LHRH)受体的器官(垂体、卵巢)。血浆蛋白结合率为86%。该药物从大多数组织中迅速清除,主要在48小时内清除完毕。 …西曲瑞克少量可通过胎盘。尚未研究西曲瑞克或其代谢物在母乳中的分布。西曲瑞克以原形经尿液排出,并在胆汁中经肽酶代谢。…健康志愿者的研究表明,西曲瑞克在人、大鼠和犬体内的排泄情况相似。皮下注射后,男性和女性的西曲瑞克绝对生物利用度均约为85%。女性的表观分布容积为1.16 ± 0.29 L/kg,男性为1.02 ± 0.33 L/kg。静脉注射后末端半衰期约为10小时,皮下注射后约为30小时,女性的末端半衰期呈下降趋势。人血浆蛋白结合率约为85%。单次(0.25、0.5 和 1.00 mg)和多次(0.25 至 1.00 mg)给药后均观察到线性药代动力学。在 3 mg 剂量范围内,药代动力学呈线性。 代谢/代谢物体外研究表明,西曲雷克斯在 I 期和 II 期代谢中均稳定。西曲雷克斯可被肽酶转化,肽(1-4)是主要代谢物。 在大鼠胆汁中,西曲雷克斯的主要代谢物被鉴定为七肽(1-7)。该代谢物在大鼠中无药理活性,即不抑制睾酮分泌。 向雄性和雌性大鼠皮下注射10 mg西曲瑞克后,24小时内即可在胆汁样本中检测到西曲瑞克以及痕量的肽(1-9)、(1-7)、(1-6)和(1-4)。体外研究表明,西曲瑞克在I期和II期代谢中均稳定。西曲瑞克经肽酶转化,其中(1-4)肽是主要代谢产物。 生物半衰期 ~62.8 小时 在人体中,静脉注射和皮下注射后的终末半衰期分别为 8-9 小时和 24-40 小时。 在大鼠中,静脉注射和皮下注射后的终末半衰期分别为 1-2 小时和 7-14 小时……消除半衰期:单次 3 mg 剂量:62.8 小时(38.2–108 小时);单次 0.25 mg 剂量:5.0 小时(2.4–48.8 小时);每日 0.25 mg,持续 14 天:20.6 小时(4.1–179.3 小时)/摘自表格/ 在排泄器官(肝脏、肾脏)、脾脏和含有 LHRH 结合位点的器官中观察到半衰期大于或等于 100 小时。 西曲瑞克 皮下注射后数小时内即可立即抑制促性腺激素(LH 比 FSH 更明显);其作用完全可逆,且呈剂量依赖性 [2]。 |
| 毒性/毒理 (Toxicokinetics/TK) |
蛋白结合率为 86%。非人毒性值为 68.1 mg/kg,被确定为最小致死剂量。临床研究表明,西曲瑞克无全身毒性和致畸性。偶见注射部位出现短期红斑,但无风团或瘙痒[2]。
|
| 参考文献 |
[1]. Reprod Biol Endocrinol.2003 Oct 7;1:65;
|
| 其他信息 |
治疗用途
西曲瑞克适用于抑制接受控制性卵巢刺激的女性出现过早的黄体生成素 (LH) 峰值。这项随机、安慰剂对照、单盲研究纳入了 45 只成年雌性 Wistar 大鼠……将子宫内膜组织植入腹腔后,大鼠被随机分为三个等量的干预组:(i) 对照组,(ii) 亮丙瑞林组,以及 (iii) 西曲瑞克组。六周后,通过第二次剖腹手术测量植入体积(体积-1)。随后,对照组每周皮下注射生理盐水(0.1 mL/只大鼠),亮丙瑞林组每日两次皮下注射亮丙瑞林(0.075 mg/kg),西曲瑞克组每日皮下注射西曲瑞克(0.001 mg/只大鼠),持续8周。治疗结束后,通过第三次剖腹手术再次测量植入物体积(体积-2),并将植入物完全取出进行组织病理学检查。比较各组内体积-1和体积-2的值,以及组间间质组织和腺体组织的评分。亮丙瑞林组和西曲瑞克组的体积-2均较体积-1显著减小(分别为P < 0.01和P < 0.01),而对照组的体积无显著变化(P > 0.05)。与对照组相比,对照组的腺体组织和间质组织均显著减少(分别为 P < 0.01 和 P < 0.01)。亮丙瑞林和西曲瑞克在缩小实验性子宫内膜异位症病灶的大小和组织学结构方面显示出相似的疗效。药物警告:西曲瑞克应由具有生育治疗经验的医护人员处方。开始使用醋酸西曲瑞克治疗前必须排除妊娠。 接受控制性卵巢刺激的患者中,1-2%报告肝功能检查结果升高,包括ALT(SGPT)、AST(SGOT)、γ-谷氨酰转移酶(GGT、GGTTP)和碱性磷酸酶,最高可达正常值上限的3倍。 对GnRH过敏的患者应谨慎使用。这些患者在首次注射后应密切监测。在一项与不孕症无关的适应症研究中,一名患者在接受西曲瑞克10毫克/天治疗7个月后出现严重的过敏反应,表现为咳嗽、皮疹和低血压。 已有局部反应(例如,发红、红斑、瘀斑、瘙痒、肿胀和瘙痒)的报告。这些不良反应通常是短暂的、轻微的且持续时间短。 有关西曲瑞克药物警告的更完整数据(共8项警告),请访问HSDB记录页面。 药效学 西曲瑞克是一种合成的十肽,具有促性腺激素释放激素 (GnRH) 拮抗活性。GnRH诱导垂体前叶促性腺激素细胞产生并释放黄体生成素 (LH) 和卵泡刺激素 (FSH)。月经周期中期,雌二醇 (E2) 的正反馈增强了促性腺激素释放激素 (GnRH) 的释放,导致黄体生成素 (LH) 峰值。LH 峰值诱导优势卵泡排卵,卵母细胞恢复减数分裂,随后发生黄体化,表现为孕酮水平升高。西曲瑞克与天然 GnRH 竞争性结合垂体细胞膜受体,从而以剂量依赖的方式控制 LH 和卵泡刺激素 (FSH) 的释放。西曲瑞克是一种 GnRH 拮抗剂,它竞争性抑制 GnRH 受体,从而抑制 LH 和 FSH 的分泌。它已被用于体外受精 (IVF) 中以预防过早的 LH 峰值 [2],并在动物研究中用于保护卵巢免受环磷酰胺引起的损伤 [3]。在人类卵巢癌细胞中,西曲瑞克在大多数细胞系(EFO-27 除外)中表现出与 GnRH-I 激动剂相当的直接抗增殖作用,并且这些作用在 GnRH-I 受体敲低后仍然存在,表明它们并非通过 GnRH-I 受体介导 [1]。在 ES-2 卵巢癌细胞中,西曲瑞克仅在浓度为 1000 ng/ml 时抑制细胞生长 [1]。 |
| 分子式 |
C70H92CLN17O14
|
|---|---|
| 分子量 |
1431.061
|
| 精确质量 |
1429.669
|
| CAS号 |
120287-85-6
|
| 相关CAS号 |
Cetrorelix Acetate;145672-81-7;Cetrorelix diacetate;130143-01-0
|
| PubChem CID |
25074887
|
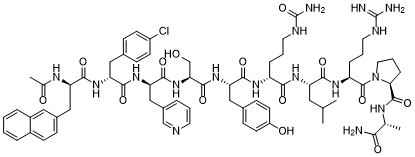
| 序列 |
Ac-D-2-Nal-D-Phe(4-Cl)-β-(3-pyridyl)-D-Ala-Ser-Tyr-D-Cit-Leu-Arg-Pro-D-Ala-NH2|Ac-D-2-Nal-D-Phe(4-Cl)-β-(3-pyridyl)-D-Ala-Ser-Tyr-D-Cit-Leu-Arg-Pro-D-Ala-NH2
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.4±0.1 g/cm3
|
| 折射率 |
1.668
|
| LogP |
2.69
|
| tPSA |
495.67
|
| 氢键供体(HBD)数目 |
16
|
| 氢键受体(HBA)数目 |
16
|
| 可旋转键数目(RBC) |
38
|
| 重原子数目 |
102
|
| 分子复杂度/Complexity |
2840
|
| 定义原子立体中心数目 |
10
|
| SMILES |
CC(C[C@H](NC([C@H](NC([C@@H](NC([C@@H](NC([C@H](NC([C@H](NC([C@H](NC(C)=O)CC1=CC2=CC=CC=C2C=C1)=O)CC3=CC=C(Cl)C=C3)=O)CC4=CN=CC=C4)=O)CO)=O)CC5=CC=C(O)C=C5)=O)CCCNC(N)=O)=O)C(N[C@H](C(N6CCC[C@H]6C(N[C@@H](C(N)=O)C)=O)=O)CCCNC(N)=N)=O)C
|
| InChi Key |
SBNPWPIBESPSIF-MHWMIDJBSA-N
|
| InChi Code |
InChI=1S/C70H92ClN17O14/c1-39(2)31-52(61(94)82-51(15-9-28-77-69(73)74)68(101)88-30-10-16-58(88)67(100)79-40(3)59(72)92)83-60(93)50(14-8-29-78-70(75)102)81-63(96)54(34-43-20-25-49(91)26-21-43)86-66(99)57(38-89)87-65(98)56(36-45-11-7-27-76-37-45)85-64(97)55(33-42-18-23-48(71)24-19-42)84-62(95)53(80-41(4)90)35-44-17-22-46-12-5-6-13-47(46)32-44/h5-7,11-13,17-27,32,37,39-40,50-58,89,91H,8-10,14-16,28-31,33-36,38H2,1-4H3,(H2,72,92)(H,79,100)(H,80,90)(H,81,96)(H,82,94)(H,83,93)(H,84,95)(H,85,97)(H,86,99)(H,87,98)(H4,73,74,77)(H3,75,78,102)/t40-,50-,51+,52+,53-,54+,55-,56-,57+,58+/m1/s1
|
| 化学名 |
(S)-1-(((R)-2-((S)-2-((S)-2-((R)-2-((R)-2-((R)-2-acetamido-3-(naphthalen-2-yl)propanamido)-3-(4-chlorophenyl)propanamido)-3-(pyridin-3-yl)propanamido)-3-hydroxypropanamido)-3-(4-hydroxyphenyl)propanamido)-5-ureidopentanoyl)-L-leucyl-L-arginyl)-N-((R)-1-amino-1-oxopropan-2-yl)pyrrolidine-2-carboxamide
|
| 别名 |
CD 20761 D-20761 D20761NS-75A NS 75A SB-075 acetate
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.6988 mL | 3.4939 mL | 6.9878 mL | |
| 5 mM | 0.1398 mL | 0.6988 mL | 1.3976 mL | |
| 10 mM | 0.0699 mL | 0.3494 mL | 0.6988 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT05951400
Conditions:IVF|PCOLink: https://clinicaltrials.gov/ct2/show/NCT06378268
Conditions:Progestins Primed Ovarian StimulationLink: https://clinicaltrials.gov/ct2/show/NCT03680053
Conditions:Infertility|ART
Title:PPOS Protocol Versus GnRH Anatagonist for Expected Normal Responder Patients Undergoing ART : a Randomized Clinical Trial
Status:Completed
updateDate:2025-03-11
Ctid:NCT06868576
Link: https://clinicaltrials.gov/ct2/show/NCT06868576
Conditions:PPOS|GnRH Antagonist|Assisted Reproductive TechniquesLink: https://clinicaltrials.gov/ct2/show/NCT06608186
Conditions:PCOS (Polycystic Ovary Syndrome)Link: https://clinicaltrials.gov/ct2/show/NCT02478775
Conditions:Obesity|FertilityLink: https://clinicaltrials.gov/ct2/show/NCT04671966
Conditions:Heart Diseases|Left Ventricular DysfunctionLink: https://clinicaltrials.gov/ct2/show/NCT04724486
Conditions:In Vitro Fertilization|Intracytoplasmic Sperm Injection|InfertilityLink: https://clinicaltrials.gov/ct2/show/NCT04724343
Conditions:In Vitro Fertilization|Intracytoplasmic Sperm Injection|InfertilityLink: https://clinicaltrials.gov/ct2/show/NCT04727671
Conditions:In Vitro Fertilization|Intracytoplasmic Sperm Injection|Infertility|Polycystic Ovary SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT04727684
Conditions:In Vitro Fertilization|Infertility|Intracytoplasmic Sperm Injection|Polycystic Ovary SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT05738382
Conditions:Assisted Reproductive Technology|Controlled Ovarian HyperstimulationLink: https://clinicaltrials.gov/ct2/show/NCT05939284
Conditions:PCOSLink: https://clinicaltrials.gov/ct2/show/NCT05751681
Conditions:Ovary Cyst|Fertility IssuesLink: https://clinicaltrials.gov/ct2/show/NCT05112692
Conditions:PCOS (Polycystic Ovary Syndrome) of Bilateral OvariesLink: https://clinicaltrials.gov/ct2/show/NCT04094467
Conditions:Polycystic Ovary SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT04308343
Conditions:Ectopic PregnancyLink: https://clinicaltrials.gov/ct2/show/NCT02042196
Conditions:Menopause|AgingLink: https://clinicaltrials.gov/ct2/show/NCT00449150
Conditions:Benign Prostatic HypertrophyLink: https://clinicaltrials.gov/ct2/show/NCT03118830
Conditions:Invitro FertilizationLink: https://clinicaltrials.gov/ct2/show/NCT02823080
Conditions:InfertilityLink: https://clinicaltrials.gov/ct2/show/NCT01457703
Conditions:ObesityLink: https://clinicaltrials.gov/ct2/show/NCT02784457
Conditions:Assisted ReproductionLink: https://clinicaltrials.gov/ct2/show/NCT01709942
Conditions:PCOS|OHSS|INFERTILITYLink: https://clinicaltrials.gov/ct2/show/NCT01109888
Conditions:Administration of Increased Dose of GnRH Antagonist for Coasting for Decreasing the Risk for Ovarian Hyperstimulation Syndrome( OHSS)Link: https://clinicaltrials.gov/ct2/show/NCT02333253
Conditions:Other Complications Associated With Artificial FertilizationLink: https://clinicaltrials.gov/ct2/show/NCT02392520
Conditions:Ovarian Hyperstimulation SyndromeLink: https://clinicaltrials.gov/ct2/show/NCT00866034
Conditions:In Vitro Fertilization|Intracytoplasmic Sperm InjectionLink: https://clinicaltrials.gov/ct2/show/NCT01225835
Conditions:InfertilityLink: https://clinicaltrials.gov/ct2/show/NCT00667758
Conditions:Rheumatoid ArthritisLink: https://clinicaltrials.gov/ct2/show/NCT01468441
Conditions:InfertilityLink: https://clinicaltrials.gov/ct2/show/NCT01595334
Conditions:InfertilityLink: https://clinicaltrials.gov/ct2/show/NCT00439829
Conditions:InfertilityLink: https://clinicaltrials.gov/ct2/show/NCT01005784
Conditions:SubfertilityLink: https://clinicaltrials.gov/ct2/show/NCT00244452
Conditions:EndometriosisLink: https://clinicaltrials.gov/ct2/show/NCT00628121
Conditions:PremenopauseLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2009-012847-40
Condition:SubfertilityLink: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-004865-17
Condition:Benign Prostatic Hyperplasia (BPH)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-000212-89
Condition:Comparar los resultados obtenidos con el empleo de antagonistas de la GnRH en la sincronización receptora de ovocitos-donante frente a los resultados obtenidos con el tradicional empleo de supresión hipofisaria con análogos de la GnRHa.To compare outcomes using gNRH antagonists versus GnRH analogues in endometrial syncronization bettwenn ovum donors and recipients in a egg donation programme.Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-002598-30
Condition:Benign Prostatic Hyperplasia (PBH)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2007-003414-34
Condition:Benign Prostatic Hyperplasia (PBH)Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2006-004460-31
Condition:Poor ovarian response in women undergoing In vitro Fertilisation (IVF) treatment.Link: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2005-003069-18
Condition:infertilitetInvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
