| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 体外研究 (In Vitro) |
通过测定母体化合物消失率测定了地西泮、氯噻西泮、托非索泮、依替唑仑、坦度螺酮和丙咪嗪六种精神药物在14种重组人肝细胞色素P450(CYP)异构体和人肝微粒体中的代谢活性。体外动力学研究表明,在人肝微粒体中,托非索泮的Vmax/Km值最高,其次为坦度螺酮>氯噻西泮>丙咪嗪、地西泮和依替唑仑。在重组CYP中,除氯噻西泮和丙咪嗪外,CYP3A4代谢活性最高。氯噻西泮的代谢由CYP2B6、CYP3A4、CYP2C18和CYP2C19催化,丙咪嗪的代谢效率最高,由CYP2D6催化。此外,地西泮、氯噻西泮和依替唑仑在人肝微粒体中的代谢活性分别被CYP3A4抑制剂2.5μM酮康唑抑制97.5%、65.1%和83.5%,而加入CYP2D6抑制剂1或10μM奎尼丁后未检测到丙咪嗪的代谢。这些结果表明,除丙咪嗪的代谢由CYP2D6催化外,所研究的精神药物主要由CYP3A4代谢。此外,这种基于消失率的方法似乎有助于识别负责旧药物的 CYP 异构体,因为尚未报道过这些旧药物的代谢特征。[1]
|
|---|---|
| 药代性质 (ADME/PK) |
Absorption, Distribution and Excretion
Etizolam is well absorbed from the intestines with a biological bioavailability of 93% following oral administration. After a single oral dosing of 0.5mg etizolam, it takes approximately 0.9 hours to reach the peak plasma concentration of 8.3 ng/mL. In a rat study, the amounts of etizolam excreted was 30% in urine was 70% in feces, while the values in a mouse study were 40% in urine and 60% in feces. Apparent distribution volume was 0.9 ± 0.2 L/kg following a single oral doing of 0.5mg etizolam. Metabolism / Metabolites Biotransformation of etizolam is extensive and involves hydroxylation and conjugation. The main metabolite formed via 1'-hydroxylation is α-hydroxyetizolam which retains pharmacological activity comparable to that of the parent drug, indicating that the action of metabolites may contribute to the clinical effects of etizolam. CYP3A4 is predicted to be the main CYP enzyme responsible for mediating etizolam metabolism. CYP2C18 and CYP2C19 are also involved in the metabolic pathways. Etizolam has known human metabolites that include 7-(2-chlorophenyl)-4-ethyl-13-methyl-3-thia-1,8,11,12-tetrazatricyclo[8.3.0.02,6]trideca-2(6),4,7,10,12-pentaen-9-ol and alpha-Hydroxyetizolam. Biological Half-Life The average elimination half life of etizolam following a single oral dose of 0.5mg is 3.4 hours but may be increased up to 17 hours depending on the rate of metabolism. The main metabolite α-hydroxyetizolam displays a longer elimination half life of 8.2 hours. |
| 毒性/毒理 (Toxicokinetics/TK) |
Toxicity Summary
Benzodiazepines bind nonspecifically to benzodiazepine receptors BNZ1, which mediates sleep, and BNZ2, which affects affects muscle relaxation, anticonvulsant activity, motor coordination, and memory. As benzodiazepine receptors are thought to be coupled to gamma-aminobutyric acid-A (GABAA) receptors, this enhances the effects of GABA by increasing GABA affinity for the GABA receptor. Binding of the inhibitory neurotransmitter GABA to the site opens the chloride channel, resulting in a hyperpolarized cell membrane that prevents further excitation of the cell. Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Etizolam is not approved for marketing in the United States by the U.S. Food and Drug Administration. Very little information is available on the passage of etizolam into milk. An alternate drug is preferred, especially while nursing a newborn or preterm infant. If etizolam is used, monitor the infant for sedation, poor feeding and poor weight gain. ◉ Effects in Breastfed Infants A woman took etizolam 1 mg and trazodone 50 mg once daily for 3 months postpartum. Her infant was over 50% breastfed and demonstrated no adverse reactions at the 1- and 3-month checkups. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. |
| 参考文献 |
Biol Pharm Bull.2005 Sep;28(9):1711-6;Curr Drug Metab.2008 Oct;9(8):827-44.
|
| 其他信息 |
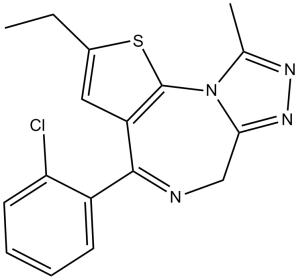
Etizolam is an organic molecular entity.
4-(2-chlorophenyl)-2-ethyl-9-methyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine, its salts, isomers, and salts of isomers is a DEA Schedule I controlled substance. Substances in the DEA Schedule I have no currently accepted medical use in the United States, a lack of accepted safety for use under medical supervision, and a high potential for abuse. It is a Temporary listing of substances subject to emergency scheduling substance. Etizolam is a thienodiazepine which is chemically related to benzodiazepine (BDZ) drug class; it differs from BDZs in having a benzene ring replaced with a thiophene ring. It is an agonist at GABA-A receptors and possesses amnesic, anxiolytic, anticonvulsant, hypnotic, sedative and skeletal muscle relaxant properties. Initially introduced in 1983 in Japan as treatment for neurological conditions such as anxiety and sleep disorders, etizolam is marketed in Japan, Italy and India. It is not approved for use by FDA in the US; however it remains unscheduled in several states and is legal for research purposes. According to the Italian P.I. sheet etizolam belongs to a new class of diazepines, thienotriazolodiazepines. This new class is easily oxidized, rapidly metabolized, and has a lower risk of accumulation, even after prolonged treatment. Etizolam has an anxiolytic action about 6 times greater than that of diazepam. Etizolam produces, especially at higher dosages, a reduction in time taken to fall asleep, an increase in total sleep time and a reduction in the number of awakenings. During tests there were not substantial changes in deep sleep. There is a reduction of REM sleep. In EEG tests of healthy volunteers Etizolam showed some characteristics of tricyclic antidepressants. Etizolam (marketed under the brand name Etilaam, Etizola, Sedekopan, Pasaden or Depas) is a thienodiazepine drug which is a benzodiazepine analog. The etizolam molecule differs from a benzodiazepine in that the benzene ring has been replaced by a thiophene ring. It possesses amnesic, anxiolytic, anticonvulsant, hypnotic, sedative and skeletal muscle relaxant properties. Drug Indication Indicated for the treatment of generalized anxiety disorder with depression, panic disorder and insomnia. Mechanism of Action Etizolam is selectively a full agonist at GABA-A receptors to increase GABAergic transmission and enhance GABA-induced Cl- currents. It is reported to bind to the benzodiazepine binding site which is located across the interface between the alpha and gamma subunits. Benzodiazapines are reported to only bind to receptors that contain gamma 2 and alpha 1/2/3/5 subunits. Alpha-1-containing receptors mediate the sedative effects of etizolam whereas alpha-2 and alpha-3 subunit-containing receptors mediate the anxiolytic effect. Etizolam shows high potency and affinity towards GABA-A receptor with alpha 1 beta 2 gamma 2S subunit combination. By binding to the regulatory site of the receptor, etizolam potentiates GABA transmission by facilitating the opening of GABA-induced chloride channels. Etizolam is a specific antagonist at PAFR. It inhibits PAF-induced platelet aggregation by inhibiting PAF binding to the receptors located on the surface of platelets with an IC50 of 22nM. Pharmacodynamics Etizolam is a CNS depressant with anxiolytic, anticonvulsant, sedative-hypnotic and muscle relaxant effects. It acts on the benzodiazepine site of the GABA-A receptor as an agonist to increase inhibitory GABAergic transmission throughout the central nervous system. Studies indicate that etizolam mediates its pharmacological actions with 6 to 10 times more potency than that of diazepam. Clinical human studies performed in Italy showed clinical effectiveness of etizolam in relieving symptoms in patients with generalized anxiety disorders with depressive symptoms. Etizolam also mediates imipramine-like neuropharmacological and behavioral effects, as well as minor effects on cognitive functioning. It is shown to substitute the actions of a short-acting barbiturate, pentobarbitol, in a drug discrimination study. Etizolam is an antagonist at platelet-activating-factor (PAF) receptor and attenuates the recurrence of chronic subdural hematoma after neurosurgery in clinical studies. It is shown to inhibit PAF-induced bronchoconstriction and hypotension. |
| 分子式 |
C17H15CLN4S
|
|
|---|---|---|
| 精确质量 |
342.07
|
|
| CAS号 |
40054-69-1
|
|
| 相关CAS号 |
|
|
| PubChem CID |
3307
|
|
| 外观&性状 |
Typically exists as solid at room temperature
|
|
| 熔点 |
147-148
|
|
| LogP |
2.6
|
|
| tPSA |
71.3
|
|
| 氢键供体(HBD)数目 |
0
|
|
| 氢键受体(HBA)数目 |
4
|
|
| 可旋转键数目(RBC) |
2
|
|
| 重原子数目 |
23
|
|
| 分子复杂度/Complexity |
474
|
|
| 定义原子立体中心数目 |
0
|
|
| InChi Key |
VMZUTJCNQWMAGF-UHFFFAOYSA-N
|
|
| InChi Code |
InChI=1S/C17H15ClN4S/c1-3-11-8-13-16(12-6-4-5-7-14(12)18)19-9-15-21-20-10(2)22(15)17(13)23-11/h4-8H,3,9H2,1-2H3
|
|
| 化学名 |
7-(2-Chlorophenyl)-4-ethyl-13-methyl-3-thia-1,8,11,12-tetraazatricyclo[8.3.0.02,6]trideca-2(6),4,7,10,12-pentaene
|
|
| 别名 |
|
|
| HS Tariff Code |
2934.99.9001
|
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
|
|||
|---|---|---|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Cetiedil
Cetiedil
 Tinlarebant
Tinlarebant
 Amelenodor (NX-13)
Amelenodor (NX-13)
 Mebufotenin
Mebufotenin
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1





























 COA
COA
