| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |

| 靶点 |
β-lactamases
|
|---|---|
| 体外研究 (In Vitro) |
ETX0282是一种口服β-内酰胺酶抑制剂(BLI)和头孢菌素前药,对a、C和D类丝氨酸β-内酰酶具有广谱活性[1]。
在R3甲基系列中,化合物23(ETX0282)的稳定性明显优于密切相关的类似物20、21和22。虽然我们不完全了解这一显著特征的原因,但我们假设化合物23采用了独特的构象和轨道重叠。这种构象可以转化为酯羰基周围的差异化立体环境,并可能影响sp2碳的亲电性[1]。 |
| 体内研究 (In Vivo) |
口服前药ETX0282的体内分析[1]
在大鼠(N=3/只)中测定了ETX1317及其异丙酯前药ETX0282的静脉内(iv)和口服(po)药代动力学。表7总结了药代动力学参数。中等清除率(CL)和低分布体积(Vdss)导致半衰期短至0.4小时,这与DBO类别的其他成员一致。13近60%的静脉注射剂量的ETX1317作为未改变的药物在尿液中消除,表明肾排泄是主要的清除机制。以10mg/kg当量剂量口服ETX0282导致ETX1317的高暴露(Cmax=5.8μg/mL和AUC=7.0μg·h/mL),没有观察到ETX0282的循环浓度,表明该物种的ETX0282迅速转化为ETX1317,生物利用度(F)很高。对ETX0282的体内外代谢进行了研究。除了ETX0282的酯前药裂解产生ETX1317外,还观察到DBO核心的水解裂解产生了ETX1317和ETX0282二胺代谢物(图S1,支持信息)。合成了ETX1317的二胺代谢物,并将其用作最终毒理学研究中量化代谢物暴露的标准。我们还寻找了N-O键断裂产生单氟乙酸酯的证据,并且在体外和体内血浆和肝组织样本中都没有观察到这些代谢物或潜在的相关结构。ETX1317的大鼠蛋白结合率较低,在5-100μM的浓度范围内,未结合的平均分数为0.91(表7)。由于ETX0282的相关性有限,因此未确定其蛋白结合。 为了支持确定ETX1317和头孢泊肟组合的PK/PD,在中性粒细胞减少的小鼠大腿感染模型32中,使用大肠杆菌MDR临床分离株ARC2687评估了ETX0282在口服给药后恢复头孢泊肟酯体内活性的能力。虽然ETX0282和头孢泊肟酯(CPDP)联合使用的最终临床适应症是治疗复杂的尿路感染(cUTI),但小鼠中性粒细胞减少性大腿模型是用于组织和非基于问题的感染的暴露效果PK/PD分析的标准模型。33该模型中确定的PK/PD暴露终点的实现已被证明与临床成功相关。34分离株ARC2687对氟喹诺酮类药物(左氧氟沙星的MIC>4μg/mL)和头孢菌素类药物(头孢泊肟的MIC>64μg/mL)耐药,但对美罗培南(MIC=0.03μg/mL)及CPD敏感:ETX1317组合(MIC=0.25μg/mL)。对于所有手臂,每只大腿接种三只CD-1雌性小鼠,每只动物有2个数据点。24小时后,与初始接种物相比,载体对照显示菌落形成单位(CFU)计数大幅增加(+4.26 log(CFU/g)),证实了该分离物的毒力(图3)。正如根据其MIC所预期的那样,单独使用CPDP(po,q6h)无效,而美罗培南(与预处理相比为-1.46 log(CFU/g))。每6小时口服一次CPDP和ETX0282的组合后,观察到所有三种剂量的BLI的CFU计数均显著下降。剂量分别为50mg/kg和100mg/kg(po,q6h)的CPDP和ET X0282分别减少了1.11log(CFU/g)[1]。 |
| 酶活实验 |
酶抑制试验[1]
用于测量β-内酰胺酶抑制的方法与Durand-Reville等人的补充中所述的方法相同。13使用Spectramax吸光板读数器在环境温度下测量了0.1 M磷酸钠(pH 7.0)、10 mM NaHCO3和0.005%Triton X-100中不同抑制剂浓度下,产色β-内酰酶底物硝基头孢在100μM下水解490 nm的反应进程曲线。这组12条进展曲线与酶失活的二阶动力学模型进行了全局拟合。使用最佳拟合曲线集上的10分钟和60分钟时间点通过Hill方程的非线性回归计算IC50:%抑制=100[I]n/(IC50n+[I]n),其中[I]是抑制剂浓度,n是Hill系数。如前所述,使用Global Kinetic Explorer根据一组进度曲线计算二阶失活速率常数kinact/Ki 根据Shapiro等人发表的方法测量大肠杆菌PBP2的抑制作用。38从溶解在由0.1 M磷酸钠(pH 6.0)和0.01%Triton X-100组成的测定缓冲液中的化合物的新鲜溶液中制备每种抑制剂的2倍系列稀释液,从61.44到0.06和0μM。将抑制剂溶液(2μL)和2μL 90 nM 5-TAMRA-氨苄青霉素加入到低容量、浅孔、黑色聚苯乙烯、384孔微孔板的孔中。通过加入2μL 300 nM大肠杆菌PBP2引发反应。反应后荧光各向异性发生变化。使用Global Kinetic Explorer从12条进度曲线中计算出二阶失活速率常数kinact/Ki。 抗菌药物敏感性试验[1] 根据CLSI39,使用CAMHB进行肉汤微量稀释敏感性试验。Durlobactam和avibactam由一家CRO公司合成。使用头孢泊肟。通过以固定的1:2重量比滴定2倍稀释的头孢泊肟与ETX1317组合,测试头孢泊肟联合ETX1317对耐多药肠杆菌临床分离株的最小抑菌浓度(MIC)。 β-内酰胺酶抑制的生化评估[2] 如Durand-Reville等人所述,在环境温度下,在0.1 M磷酸钠(pH 7.0)、10 mM NaHCO3和0.005%Triton X-100的抑制剂浓度范围内,测量了每种酶在490 nm下水解100μM硝基头孢的反应进展曲线。这组曲线全局拟合到酶失活的二阶动力学模型。使用最佳拟合曲线集上的60分钟时间点通过Hill方程的非线性回归计算IC50:%抑制=100[I]n/(IC50+[I]n),其中[I]是抑制剂浓度,n是Hill系数。 青霉素结合蛋白的BOILLIN标记和与ETX1317或对照化合物的竞争[2] 将化合物溶解并稀释在0.1 M磷酸钠(pH 7.0)中,在30°C下与5 mg/mL大肠杆菌膜一起孵育30分钟。将BOCILLIN FL加入至终浓度为120μM,并继续孵育30分钟。将N-月桂酰arcosine加入至终含量为2%(w/v),以溶解含有PBPs的内膜。将样品在16000g下离心10分钟,使外膜颗粒化。上清液(10μL)与4μL TruPAGE LDS样品缓冲液和2μL 10×TruPAGE样品还原剂混合。样品在70°C下加热20分钟,然后在4-20%丙烯酰胺TruPAGE梯度凝胶上用TruPAGE TEA tricine运行缓冲液在140 V下电泳2.5小时。凝胶用水短暂洗涤,在50%甲醇/10%乙酸/40%水中固定15分钟,用水洗涤1小时。使用绿色荧光通道用Azure Biosystems C600凝胶成像仪拍摄BOCILLIN FL荧光,并显示为阴性。 青霉素结合蛋白的BOILLIN标记以及与头孢泊肟或对照化合物的竞争。 |
| 细胞实验 |
体外DMPK实验[1]
将来自大鼠和人类组织的肝脏和肠道S9亚细胞组分在pH 7.4的100 mM磷酸钾缓冲液中稀释至0.8 mg/mL的蛋白质浓度,并在加入10μM(最终)目标化合物之前在37°C水浴中预孵育5分钟。在0、2、5、10、20、40和60分钟时取出连续等分试样,并在LC/MS/MS30之前用内标在乙腈中淬灭,以确定前药的浓度。根据耗竭数据的对数线性图的斜率确定了一阶退化半衰期。 使用相同的方案评估缓冲液稳定性(磷酸钾缓冲液,pH 7.4),但不添加S9亚细胞组分。 抗生素敏感性试验和数据分析[2] 根据CLSI指南,使用CAMHB进行肉汤微量稀释敏感性测试。ETX1317和阿维巴坦在CRO中合成。使用头孢他啶、哌拉西林、他唑巴坦、阿莫西林、头孢克肟、头孢呋辛、头孢泊肟和氯雷他定。通过以固定的1:2重量比滴定头孢泊肟与ETX1317组合的2倍稀释液来测试头孢泊肟联合ETX1317的最小抑菌浓度(MIC)。在固定为4μg/mL的阿维巴坦存在下,通过滴定头孢他啶的2倍系列稀释液来测试头孢他啶-阿维巴旦的MIC,在固定为3μg/mL的他唑巴坦存在下,滴定哌拉西林的2倍序列稀释液来检测哌拉西林-他唑巴坦的MIC。对于SENTRY监测研究,在爱荷华州北自由的JMI实验室进行了参考肉汤微稀释敏感性试验,使用JMI实验室根据临床和实验室标准研究所指南使用CAMHB从新鲜制备的库存中提取的96孔冷冻板。SENTRY监测数据库提供了其他对照药物(如有)的敏感性数据,包括阿莫西林-克拉维酸(2:1)、头孢他啶、左氧氟沙星、美罗培南、呋喃妥因、哌拉西林-他唑巴坦和甲氧苄啶-磺胺甲恶唑。根据CLSI M100(2018)指南进行质量控制(QC)和结果解释。 大肠杆菌膜的制备[2] 大肠杆菌W3110ΔampC(缺乏β-内酰胺酶活性)在37°C下在2升CAMHB中生长至OD600=0.6。在4°C下以5000g离心15分钟收集细胞。颗粒储存在-80°C下。将冷冻颗粒重新悬浮在45 mL冰冷的0.1 M磷酸钠(pH 7.0)中,并在4°C下以5000g离心10分钟。将沉淀物重新悬浮在20mL冰冷的0.1M磷酸钠(pH 7.0)中,该磷酸钠含有一种不含EDTA的蛋白酶抑制剂鸡尾酒迷你片、2mg DNase 1和10mM MgSO4。提取物在4°C下以18000 psi的压力通过法国压榨机两次,然后在4°C下以12000g的压力离心10分钟。用冷的0.1 M磷酸钠(pH 7.0)将上清液体积增加到40 mL,并在4°C下以60000 g离心80分钟。将第二个沉淀物重新悬浮在300μL 0.1 M磷酸钠(pH 7.0)中,并储存在-80°C下。通过Bradford测定法测量蛋白质浓度。 显微镜[2] 大肠杆菌ARC2687(blaAmpC,blaCTX-M-14)在35°C下在3 mL CAMHB中振荡培养过夜。受试化合物对该菌株的MIC如下:头孢泊肟>64μg/mL,ETX1317=1μg/mL,头孢泊肟-ETX317(1:2)=0.06/0.135μg/mL。细胞在CAMHB中传代1/100,在35°C下振荡至指数期(OD600≈0.3)。然后在指定浓度的化合物存在下,将培养物在CAMHB中稀释至OD600为0.02,并在35°C下振荡培养3小时。将5μL体积的培养物点样到显微镜载玻片上制备的琼脂糖垫上。加入盖玻片,使用cellSens Standard 1.12软件进行图像采集,在油下用奥林巴斯BX53显微镜在600倍放大倍数下观察细菌。 |
| 动物实验 |
In Vivo Pharmacokinetics and Pharmacology Experiments [1]
Intravenous pharmacokinetics of ETX1317 were investigated in jugular vein-cannulated male Sprague Dawley rats (Charles River Laboratories, 180–200 g, n = 3/route) at a dose of 10 mg/kg formulated in 0.9% saline. Oral pharmacokinetics of ETX0282 were evaluated at a 10 mg/kg equivalent dose of ETX1317 formulated in 25:75 PEG 400/WFI. The pH of each formulation was verified to fall within a range of 4–7, and compounds were confirmed to be in solution prior to dosing. Potency was verified by LC/MS/MS. Dose volumes were 5 and 10 mL/kg for ETX1317 (iv) and ETX0282 (po), respectively. Blood samples to be processed for plasma using K2EDTA as an anticoagulant were taken at 0.08, 0.17, 0.25, 0.5, 1, 2, 4, 8 h (iv) and 0.25, 0.5, 1, 2, 4, 8, and 24 h (po) postdose in rats. Blood samples collected into 0.5 mL of BD microtainers containing 1.0 mg of K2EDTA were centrifuged in a microfuge for 10 min. Plasma was transferred to 96-well cryotubes and stored at −80 °C prior to analysis. In vivo neutropenic infection models in the mouse were conducted as previously described.32,45 All procedures were performed to Entasis-approved IACUC policies and guidelines as well as OLAW standards. Briefly, female CD-1 mice (N = 3 per arm, 20–22 g, Charles River Laboratories) were acclimated for 5 days prior to start of study. Animals were housed 3 per cage with free access to food and water. Mice were rendered neutropenic via two doses of cyclophosphamide on days −4 and −1 with 150 mg/kg and 100 mg/kg delivered intraperitoneally in a dose volume of 10 mL/kg, respectively. E. coli isolate ARC2687 was prepared for infection from an overnight plate culture. A portion of the plate was resuspended in sterile saline and adjusted to an OD of 0.1 at 625 nm. The adjusted bacterial suspension was further diluted to target an infecting inoculum of approximately 5.0 × 106 to 1.0 × 107 CFU/mouse. The actual inoculum size varied between 5.5 × 106 and 1.6 × 107 CFU/thigh and was administered via intramuscular injection of 100 μL. Plate counts of the inoculum were performed to confirm inoculum concentration. Treatment was initiated 2 h after bacterial challenge. ETX0282 and CPDP were formulated in 0.5% HPMC/0.1% Tween 80 suspension. Meropenem was dissolved in water for injection. The in vivo efficacy study used meropenem dosed at 600 mg/kg q6h subcutaneously as positive control. For dose arms utilizing a combination of ETX0282 and CPDP, both agents were reconstituted in the same vehicle. All dose concentrations were adjusted to deliver targeted mg/kg doses within a dose volume of 10 mL/kg. Formulation potency was verified by LC/MS/MS.30 ETX0282 and CPDP were orally administered via q6h dose intervals in order to achieve targeted unbound exposures. At 24 h after initiation of therapy, animals were euthanized, and thighs (2 separate samples/animal) were aseptically collected and homogenized in 1 mL of sterile saline. Bacterial colony enumeration of tissue homogenate was performed by serial dilution on tryptic soy agar (TSA) plates, which were incubated overnight at 35 °C prior to colony-forming unit (CFU) counting. |
| 药代性质 (ADME/PK) |
The intravenous (iv) and oral (po) pharmacokinetics of ETX1317 and its isopropyl ester prodrug ETX0282 were determined in rats (N = 3/arm). Pharmacokinetic parameters are summarized in Table 7. Moderate clearance (CL) and a low volume of distribution (Vdss) resulted in a short half-life of 0.4 h which is consistent with other members of the DBO class.13 Nearly 60% of the administered iv dose of ETX1317 was eliminated in the urine as unchanged drug suggesting renal excretion was the predominant clearance mechanism. Oral administration of ETX0282 at a 10 mg/kg equivalent dose of ETX1317 resulted in high exposure of ETX1317 (Cmax = 5.8 μg/mL and AUC = 7.0 μg·h/mL) with no circulating concentrations of ETX0282 observed suggesting rapid conversion of ETX0282 to ETX1317 and high bioavailability (F) in this species. The metabolism of ETX0282 was studied in vitro and in vivo. In addition to ester prodrug cleavage of ETX0282 to yield ETX1317, hydrolytic cleavage of the DBO core was observed to yield the diamine metabolite of both ETX1317 and ETX0282 (Figure S1, Supporting Information). The diamine metabolite of ETX1317 was synthesized and used as a standard to quantify the exposure of the metabolite in definitive toxicology studies. We also looked for evidence of N–O bond cleavage to yield the monofluoro acetate and have not observed these metabolites or potentially associated structures in both in vitro and in vivo plasma and liver tissue samples. Rat protein binding of ETX1317 was low with a mean fraction unbound of 0.91 across a concentration range of 5–100 μM (Table 7). Protein binding of ETX0282 was not determined due to its limited relevance.[1]
|
| 毒性/毒理 (Toxicokinetics/TK) |
The safety of ETX0282 was investigated with a 14-day toxicology study under Good Laboratory Practice (GLP) in rats and dogs following daily oral administration. The doses of 500 mg kg–1 day–1 and 400 mg kg–1 day–1 were established as the NOAELs (no observed adverse effect level) for rats and dogs, respectively. These results support continuing development of ETX0282. Phase 1 clinical trials in healthy volunteers showed that ETX0282 was generally well-tolerated, either alone or in combination with cefpodoxime proxetil, with no serious adverse events reported (ClinicalTrials.gov identifier: NCT03491748).[1]
|
| 参考文献 |
[1]. Discovery of an Orally Available Diazabicyclooctane Inhibitor (ETX0282) of Class A, C, and D Serine β-Lactamases. J Med Chem. 2020 Jul 13;63(21):12511–12525.
[2]. In Vitro Characterization of ETX1317, a Broad-Spectrum β-Lactamase Inhibitor That Restores and Enhances β-Lactam Activity against Multi-Drug-Resistant Enterobacteriales, Including Carbapenem-Resistant Strains. ACS Infect Dis . 2020 Jun 12;6(6):1389-1397. |
| 其他信息 |
Multidrug resistant Gram-negative bacterial infections are an increasing public health threat due to rapidly rising resistance toward β-lactam antibiotics. The hydrolytic enzymes called β-lactamases are responsible for a large proportion of the resistance phenotype. β-Lactamase inhibitors (BLIs) can be administered in combination with β-lactam antibiotics to negate the action of the β-lactamases, thereby restoring activity of the β-lactam. Newly developed BLIs offer some advantage over older BLIs in terms of enzymatic spectrum but are limited to the intravenous route of administration. Reported here is a novel, orally bioavailable diazabicyclooctane (DBO) β-lactamase inhibitor. This new DBO, ETX1317, contains an endocyclic carbon–carbon double bond and a fluoroacetate activating group and exhibits broad spectrum activity against class A, C, and D serine β-lactamases. The ester prodrug of ETX1317, ETX0282, is orally bioavailable and, in combination with cefpodoxime proxetil, is currently in development as an oral therapy for multidrug resistant and carbapenem-resistant Enterobacterales infections.[1]
Multi-drug-resistant Enterobacteriales expressing a wide array of β-lactamases are emerging as a global health threat in both hospitals and communities. Although several intravenous drugs have recently been approved to address this need, there are no oral Gram-negative agents that are both safe and broadly effective against such pathogens. The lack of an effective oral agent is of concern for common infections which could otherwise be treated in the community but, due to antibiotic resistance, require hospitalization to allow for intravenous therapy. ETX1317 is a novel, broad spectrum, serine β-lactamase inhibitor of the diazabicyclooctane class that restores the antibacterial activity of multiple β-lactams against multiple species of multi-drug-resistant Enterobacteriales, including carbapenem-resistant strains. A combination of its oral prodrug, ETX0282, and the oral prodrug of a third-generation cephalosporin, cefpodoxime proxetil, is currently in clinical development. This report describes the biochemical and microbiological properties of ETX1317, which is more potent and demonstrates a greater breadth of inhibition than avibactam, the parenteral prototype of this class of β-lactamase inhibitors.[2] |
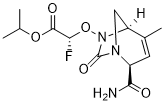
| 分子式 |
C13H18FN3O5
|
|---|---|
| 分子量 |
315.30
|
| 精确质量 |
315.123
|
| 元素分析 |
C, 49.52; H, 5.75; F, 6.03; N, 13.33; O, 25.37
|
| CAS号 |
2209871-83-8
|
| PubChem CID |
146170992
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| LogP |
0.1
|
| tPSA |
102
|
| 氢键供体(HBD)数目 |
1
|
| 氢键受体(HBA)数目 |
6
|
| 可旋转键数目(RBC) |
6
|
| 重原子数目 |
22
|
| 分子复杂度/Complexity |
536
|
| 定义原子立体中心数目 |
3
|
| SMILES |
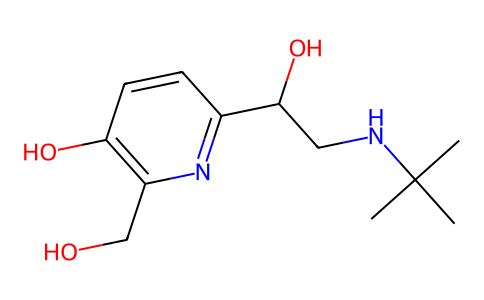
CC1=C[C@@H](N2C[C@@H]1N(C2=O)O[C@@H](C(=O)OC(C)C)F)C(=O)N
|
| InChi Key |
OMNVFPBGXYKTDB-UTLUCORTSA-N
|
| InChi Code |
InChI=1S/C13H18FN3O5/c1-6(2)21-12(19)10(14)22-17-9-5-16(13(17)20)8(11(15)18)4-7(9)3/h4,6,8-10H,5H2,1-3H3,(H2,15,18)/t8-,9+,10+/m1/s1
|
| 化学名 |
propan-2-yl (2R)-2-[[(2R,5R)-2-carbamoyl-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-en-6-yl]oxy]-2-fluoroacetate
|
| 别名 |
ETX 0282; ETX 0282; ETX0282; 2209871-83-8; GTPL10781; isopropyl (R)-2-(((1R,2R,5R)-2-carbamoyl-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-en-6-yl)oxy)-2-fluoroacetate; propan-2-yl (2R)-2-[[(2R,5R)-2-carbamoyl-4-methyl-7-oxo-1,6-diazabicyclo[3.2.1]oct-3-en-6-yl]oxy]-2-fluoroacetate; ETX0282
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1716 mL | 15.8579 mL | 31.7158 mL | |
| 5 mM | 0.6343 mL | 3.1716 mL | 6.3432 mL | |
| 10 mM | 0.3172 mL | 1.5858 mL | 3.1716 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
