| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| Other Sizes |
|

| 靶点 |
Aurora-A (IC50 = 0.031 μM)
Phthalazinone pyrazole (1 and 10 μM; 30 hours) increases the ability of HLCs to proliferate[2]. Phthalazinone pyrazole (1, 10, and 100 μM; 5 days) increases the morphological changes of the liver in differentiated HLCs without causing cytotoxicity[2]. Phthalazinone pyrazole (1 and 10 μM; 5 and 17 days) inhibits the AKT signaling pathway through an off-target effect and concurrent upregulation of HNF4α, rather than directly inhibiting Aurora-A, which suppresses the EMT and induces maturation of HLCs. Western blot and qPCR are used to confirm the outcome[2]. Phthalazinone pyrazoles are potent and selective inhibitors of Aurora-A kinase. Representative compounds such as 7 (IC₅₀ = 0.031 μM), 8 (0.029 μM), 9 (0.023 μM), 10 (0.024 μM), 11 (0.24 μM), 12 (0.093 μM), 13 (0.037 μM), 16 (0.065 μM), 17 (0.049 μM), 18 (0.025 μM), 19 (0.014 μM), 20 (0.056 μM), 21 (0.032 μM), 22 (0.036 μM), 23 (0.093 μM), 24 (0.27 μM), 25 (0.017 μM), 26 (0.109 μM), 27 (0.067 μM), 28 (0.1 μM), 29 (0.72 μM), 30 (0.071 μM), 31 (0.12 μM), 32 (0.27 μM), 33 (0.035 μM), 34 (0.115 μM), 35 (0.066 μM), and 36 (0.55 μM) showed >1000-fold selectivity over Aurora-B (Aurora-B IC₅₀ typically >100 μM for most compounds). [1] |
|---|---|
| 体外研究 (In Vitro) |
酞嗪酮吡唑(1 和 10 μM;30 小时)可增加 HLC 的增殖能力[2]。
酞嗪酮吡唑(1、10 和 100 μM;5 天)可增加分化的 HLC 中肝脏的形态变化,但不影响 HLC 的增殖能力。引起细胞毒性[2]。 Phthalazinonepyrazor(1 和 10 μM;5 和 17 天)通过脱靶效应和同时上调 HNF4α 来抑制 AKT 信号通路,而不是直接抑制 Aurora-A,Aurora-A 会抑制EMT 并诱导 HLC 成熟。 Western blot 和 qPCR 用于确认结果[2]。 该系列化合物在多种癌细胞系中表现出抗增殖活性,包括 HCT116、Colo205 和 MCF7。例如,化合物 7 在 HCT116、Colo205 和 MCF7 中的 IC₅₀ 分别为 7.8 μM、2.9 μM 和 1.6 μM。化合物 33 在 HCT116、Colo205 和 MCF7 中的 IC₅₀ 分别为 0.4 μM、1.7 μM 和 1.6 μM。部分化合物在 MCF7 细胞中表现出 Aurora-A 抑制表型,导致中心体形成不良并停滞在 G2/M 期。[1] 该系列化合物在多种癌细胞系中表现出抗增殖活性,包括 HCT116、Colo205 和 MCF7。例如,化合物 7 在 HCT116、Colo205 和 MCF7 中的 IC₅₀ 分别为 7.8 μM、2.9 μM 和 1.6 μM。化合物 33 在 HCT116、Colo205 和 MCF7 中的 IC₅₀ 分别为 0.4 μM、1.7 μM 和 1.6 μM。部分化合物在 MCF7 细胞中表现出 Aurora-A 抑制表型,导致中心体形成不良并停滞在 G2/M 期。[1] |
| 体内研究 (In Vivo) |
化合物 16 和 33 在口服给药 30 mg/kg 后,血浆浓度高于细胞 IC₅₀,Cmax 分别为 13 μM 和 25 μM。其口服生物利用度较 VX-680 显著提升。[1]
化合物 16 和 33 在口服给药 30 mg/kg 后,血浆浓度高于细胞 IC₅₀,Cmax 分别为 13 μM 和 25 μM。其口服生物利用度较 VX-680 显著提升。[1] |
| 酶活实验 |
Aurora 激酶活性通过生化激酶实验测定。将重组 Aurora-A 或 Aurora-B 激酶与测试化合物及 30 μM ATP 在含有封闭剂的激酶缓冲液中孵育。反应 40 分钟后加入 EDTA 终止。使用抗磷酸化组蛋白 H3(Ser10)抗体和 HRP 标记的二抗检测磷酸化水平,随后使用 TMB 显色。读取吸光度,并通过非线性曲线拟合计算 IC₅₀ 值。所有数据均为三次重复实验的平均值。[1]
Aurora 激酶活性通过生化激酶实验测定。将重组 Aurora-A 或 Aurora-B 激酶与测试化合物及 30 μM ATP 在含有封闭剂的激酶缓冲液中孵育。反应 40 分钟后加入 EDTA 终止。使用抗磷酸化组蛋白 H3(Ser10)抗体和 HRP 标记的二抗检测磷酸化水平,随后使用 TMB 显色。读取吸光度,并通过非线性曲线拟合计算 IC₅₀ 值。所有数据均为三次重复实验的平均值。[1] |
| 细胞实验 |
使用 CellTiter-Glo 实验评估 HCT116 细胞的活力。细胞以每孔 1000 个接种于 384 孔板中,并用测试化合物处理(浓度范围 30 μM 至 0.0015 μM,10 个浓度点,1:3 稀释)。5 天后加入 CellTiter-Glo 试剂,细胞裂解后检测发光信号。数据均为三次重复实验的平均值。[1]
使用 CellTiter-Glo 实验评估 HCT116 细胞的活力。细胞以每孔 1000 个接种于 384 孔板中,并用测试化合物处理(浓度范围 30 μM 至 0.0015 μM,10 个浓度点,1:3 稀释)。5 天后加入 CellTiter-Glo 试剂,细胞裂解后检测发光信号。数据均为三次重复实验的平均值。[1] |
| 动物实验 |
在小鼠体内进行了药代动力学研究。化合物以 30 mg/kg 的剂量口服给药。通过测量血浆浓度随时间的变化来确定 Cmax、半衰期 (T₁/₂) 和口服生物利用度。实验部分未描述具体的制剂和赋形剂信息。[1]
|
| 药代性质 (ADME/PK) |
部分化合物在微粒体和肝细胞中表现出中等至较高的稳定性。例如,化合物7的肝清除率(CLhep)为2.3 μL/min/10⁶个细胞(大鼠),微粒体清除率(CLmic)分别为0 μL/min/mg蛋白(人)和15.7 μL/min/mg蛋白(小鼠)。化合物16的CLmic分别为7.4 μL/min/mg蛋白(人)和28 μL/min/mg蛋白(小鼠)。化合物33在30 mg/kg口服给药后,半衰期为2.4小时,Cmax为25 μM。与VX-680相比,其口服生物利用度显著提高。[1]例如,化合物 7 的肝清除率 (CLhep) 为 2.3 μL/min/10⁶ 个细胞(大鼠),微粒体清除率 (CLmic) 为 0 μL/min/mg 蛋白(人)和 15.7 μL/min/mg 蛋白(小鼠)。化合物 16 的 CLmic 为 7.4 μL/min/mg 蛋白(人)和 28 μL/min/mg 蛋白(小鼠)。化合物 33 在 30 mg/kg 口服给药后,半衰期为 2.4 小时,Cmax 为 25 μM。与 VX-680 相比,其口服生物利用度显著提高。[1]
|
| 毒性/毒理 (Toxicokinetics/TK) |
在测试浓度下(例如,CYP3A4、2C9 和 2D6 的浓度 >50 μM),化合物 7、16 和 33 均未观察到明显的细胞色素 P450 抑制或 hERG 通道抑制。化合物 7 在 10 μM 时显示出 20% 的 hERG 抑制率。[1]
|
| 参考文献 |
|
| 其他信息 |
酞嗪酮吡唑是一类新型的Aurora-A抑制剂,对Aurora-B具有极高的选择性(>1000倍)。它们具有良好的口服生物利用度和血浆暴露量,使其适用于药效学研究。化合物7与Aurora-A结合的晶体结构揭示了其与铰链区的关键氢键,并解释了其对Aurora-B的选择性是由于与Aurora-B中谷氨酸残基的空间位阻所致。[1]
酞嗪酮吡唑是一类新型的Aurora-A抑制剂,对Aurora-B具有极高的选择性(>1000倍)。它们具有良好的口服生物利用度和血浆暴露量,使其适用于药效学研究。化合物 7 与 Aurora-A 结合的晶体结构揭示了其与铰链区的关键氢键,并解释了其对 Aurora-B 的选择性,这是由于与 Aurora-B 中的谷氨酸残基发生空间位阻冲突所致。[1] |
| 分子式 |
C18H15N5O
|
|---|---|
| 分子量 |
317.344602823257
|
| 精确质量 |
317.127
|
| 元素分析 |
C, 68.13; H, 4.76; N, 22.07; O, 5.04
|
| CAS号 |
880487-62-7
|
| 相关CAS号 |
880487-62-7;
|
| PubChem CID |
11652621
|
| 外观&性状 |
Brown to gray solid powder
|
| 密度 |
1.4±0.1 g/cm3
|
| 沸点 |
557.7±52.0 °C at 760 mmHg
|
| 闪点 |
291.1±30.7 °C
|
| 蒸汽压 |
0.0±1.5 mmHg at 25°C
|
| 折射率 |
1.716
|
| LogP |
3.03
|
| tPSA |
78.83
|
| 氢键供体(HBD)数目 |
2
|
| 氢键受体(HBA)数目 |
3
|
| 可旋转键数目(RBC) |
3
|
| 重原子数目 |
24
|
| 分子复杂度/Complexity |
502
|
| 定义原子立体中心数目 |
0
|
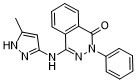
| SMILES |
O=C1N(C2C=CC=CC=2)N=C(NC2C=C(C)NN=2)C2C1=CC=CC=2
|
| InChi Key |
DSDIWWSXOOXFSI-UHFFFAOYSA-N
|
| InChi Code |
InChI=1S/C18H15N5O/c1-12-11-16(21-20-12)19-17-14-9-5-6-10-15(14)18(24)23(22-17)13-7-3-2-4-8-13/h2-11H,1H3,(H2,19,20,21,22)
|
| 化学名 |
4-[(5-methyl-1H-pyrazol-3-yl)amino]-2-phenylphthalazin-1-one
|
| 别名 |
Phthalazinone pyrazole
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: ≥ 11.11 mg/mL (~35.0 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1512 mL | 15.7560 mL | 31.5119 mL | |
| 5 mM | 0.6302 mL | 3.1512 mL | 6.3024 mL | |
| 10 mM | 0.3151 mL | 1.5756 mL | 3.1512 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 LS-Q2
LS-Q2
 VU6080195
VU6080195
 SK2188
SK2188
 Tapencarium
Tapencarium
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
