
| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| Other Sizes |
|
| 体外研究 (In Vitro) |
Zenagamtide sodium 可激活人、小鼠和大鼠细胞系统中的 GLP-1、胰淀素和降钙素受体[1]。Zenagamtide sodium(10 倍至 3 倍系列稀释;30 分钟至 3 小时)可有效激活人、小鼠和大鼠的 GLP-1R、小鼠和大鼠的 AMY3(a)R 以及小鼠和大鼠的 CTR,其 EC50 值范围为 8.4×10⁻¹³ M 至 7.8×10⁻¹¹ M[2]。Zenagamtide sodium(10 倍系列稀释;30 分钟)可有效激活人 AMY1(a)R、AMY2(a)R、AMY3(a)R 和 CTR(a),其 EC50 值范围为 1.30×10⁻¹² M 至 1.72×10⁻¹¹ M[2]。
|
|---|---|
| 体内研究 (In Vivo) |
对饮食诱导肥胖大鼠皮下注射Zenagamtide(钠)(亚慢性、慢性给药)可降低总能量摄入和体重,并改善胰岛素敏感性和代谢健康指标[1]。Zenagamtide(钠)(10 nmol/kg;皮下;单次给药)可使正常体重雄性Sprague Dawley大鼠48小时累积食物摄入量减少50%[2]。Zenagamtide(钠)(2-10 nmol/kg;皮下;每日两次;持续21天)可使雄性DIO C57Bl/6J小鼠的总食物摄入量减少23-30%,并以剂量依赖的方式诱导载体校正后的体重减轻15.8-21.3%[2]。 Zenagamtide(钠)(1→3→10 nmol/kg;皮下注射;每日一次;持续21天)可使雄性DIO Sprague Dawley大鼠的总能量摄入减少47.4%,并使体重(以载体校正后)减轻18.3%,同时维持总能量消耗[2]。Zenagamtide(钠)(1→3→10 nmol/kg;皮下注射;每日一次;持续35天)可使雄性DIO Sprague Dawley大鼠的HOMA-IR降低47%,并使稳态葡萄糖输注率增加三倍,表明胰岛素敏感性得到改善[2]。 Zenagamtide(钠)(10-30 nmol/kg;皮下注射;每日两次;持续12周)可降低血浆肝酶水平,改善55.6-64.7%小鼠的MASLD活动评分,并降低经活检证实的GAN DIO-MASH雄性C57BL/6JRj小鼠的肝脏炎症和纤维化标志物[2]。荧光标记的Zenagamtide(钠)(100 nmol/kg;静脉注射;单次给药)可到达雄性C57BL/6J小鼠参与能量摄入调节的关键脑区,包括脑室周围器官和下丘脑/后脑区域[2]。
|
| 动物实验 |
动物/疾病模型: C57Bl/6J(雄性,25周龄,体重39-56克,饮食诱导肥胖)[2]
剂量: 2 nmol/kg;10 nmol/kg 给药途径: 皮下注射;每日两次;持续21天 实验结果: 2 nmol/kg剂量组21天总食物摄入量减少23%,10 nmol/kg剂量组减少30%。2 nmol/kg剂量组和10 nmol/kg剂量组分别使载体校正后的体重减轻15.8%和21.3%,两种目标试剂剂量组之间存在显著差异。 动物/疾病模型: Sprague Dawley 小鼠(雄性,约 750 克,饮食诱导肥胖)[2] 剂量: 1 nmol/kg(第 1 天);3 nmol/kg(第 2 天);10 nmol/kg(第 3-21 天) 给药途径: 皮下注射;每日一次;持续 21 天 实验结果: 21 天后,与载体对照组相比,体重减轻了 18.3%。与载体对照组相比,总能量摄入量减少了 47.4%。与载体组相比,总能量消耗无显著差异,而限制热量摄入的体重匹配组的总能量消耗比目标试剂组低 13.6%。 动物/疾病模型: Sprague Dawley小鼠(雄性,饮食诱导肥胖)[2] 剂量: 1 nmol/kg(第1天);3 nmol/kg(第2天);10 nmol/kg(第3-35天) 给药途径: 皮下注射;每日一次;持续35天 实验结果: 空腹血糖基线值降低7%(载体组:121.1 ± 3.0 mg/dL vs. 目标试剂组:112 ± 2.1 mg/dL;p < 0.05)。空腹血浆胰岛素基线值降低42%(载体组:77.0 ± 6.7 µU/mL vs. 目标试剂组:44.8 ± 4.0 µU/mL;p < 0.001)。 HOMA-IR 降低了 47%(载体组:23.4 ± 2.3 vs. 靶试剂组:12.5 ± 1.2;p < 0.001)。钳夹试验期间,维持血糖正常所需的葡萄糖输注速率是对照组的三倍(平均稳态葡萄糖输注率:载体组:5.53 ± 1.17 mg/kg/min vs. 靶试剂组:16.4 ± 2.10 mg/kg/min;p < 0.001)。与载体对照组相比,示踪剂测定的葡萄糖摄取量增加了 37%(载体组:15.7 ± 1.51 mg/kg/min vs. 靶试剂组:24.8 ± 1.33 mg/kg/min;p < 0.001),而两组的肝葡萄糖生成量无差异。 动物/疾病模型: C57BL/6JRj(雄性,Gubra-Amylin NASH 饮食诱导的肥胖代谢功能障碍相关脂肪肝疾病,纤维化分期 F2-3,脂肪变性评分 3,炎症评分 ≥2)[2] 剂量: 10 nmol/kg(滴定:0.5→1→2→6→10 nmol/kg);30 nmol/kg(滴定:0.5→1→2→6→12→30 nmol/kg) 给药途径: 皮下注射;每日两次;12 周 实验结果: 两种剂量均显著降低了血浆丙氨酸氨基转移酶 (ALT) 和天冬氨酸氨基转移酶 (AST) 水平(与载体相比,p < 0.001)。在接受 10 nmol/kg 治疗的小鼠中,55.6% 的小鼠 MASLD 活动评分改善 ≥2 分;在接受 30 nmol/kg 治疗的小鼠中,64.7% 的小鼠 MASLD 活动评分改善 ≥2 分(与载体组相比,p < 0.001,载体组改善率为 0%)。两种剂量均显著降低了肝脏半乳糖凝集素-3 染色(炎症标志物)强度(10 nmol/kg 组 p < 0.05,30 nmol/kg 组 p < 0.01,与载体组相比)。两种剂量均显著降低了肝脏 α-平滑肌肌动蛋白染色(纤维化标志物)强度(与载体组相比,p < 0.001)。10 nmol/kg 组和 30 nmol/kg 组肝细胞中脂滴的比例分别降低至 42.2% 和 29.4%(与载体组的 77.9% 相比,p < 0.001)。 动物/疾病模型: C57BL/6J(雄性,25 g)[2] 剂量: 100 nmol/kg 给药途径: 静脉注射;单次给药 实验结果: 给药后2小时和6小时,在脑室周围器官(后区、正中隆起、终板血管器、穹窿下器)和血脑屏障保护区域(弓状核、孤束核、迷走神经背侧运动核)检测到荧光信号。给药后2小时,脑室周围器官的信号强度最高。 |
| 参考文献 |
|
| 分子式 |
C343H550N94O116.XNA
|
|---|---|
| 分子量 |
7846.60 (free base)
|
| 相关CAS号 |
Zenagamtide; 3005889-81-3
|
| 序列 |
His-Aib-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Glu-Gln-Ala-Ala-Arg-Glu-Phe-Ile-Ala-Trp-Leu-Val-Arg-Gly-Arg-{Lys(AEEA-AEEA-γGlu-C18 diacid)}-Gly-Gly-Gly-Gly-Glu-Ala-Ser-Glu-Leu-Ser-Thr-Ala-Ala-Leu-Gly-Arg-Leu-Ser-Ala-Glu-Leu-His-Glu-Leu-Ala-Thr-Leu-Pro-Arg-Thr-Glu-Thr-Gly-Ser-Gly-Ser-Pro-NH2H-Aib-EGTFTSDVSSYLEEQAAREFIAWLVRGR-{Lys(AEEA-AEEA-γGlu-C18 diacid)}-GGGGEASELSTAALGRLSAELHELATLPRTETGSGSP-NH2
|
| 短序列 |
H-Aib-EGTFTSDVSSYLEEQAAREFIAWLVRGR-{Lys(AEEA-AEEA-γGlu-C18 diacid)}-GGGGEASELSTAALGRLSAELHELATLPRTETGSGSP-NH2
|
| 外观&性状 |
White to off-white solid
|
| 别名 |
Amycretin sodium; NN 9487 sodium
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
H2O : ~10 mg/mL (with sonication)
DMSO : ~2 mg/mL (with sonication) |
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
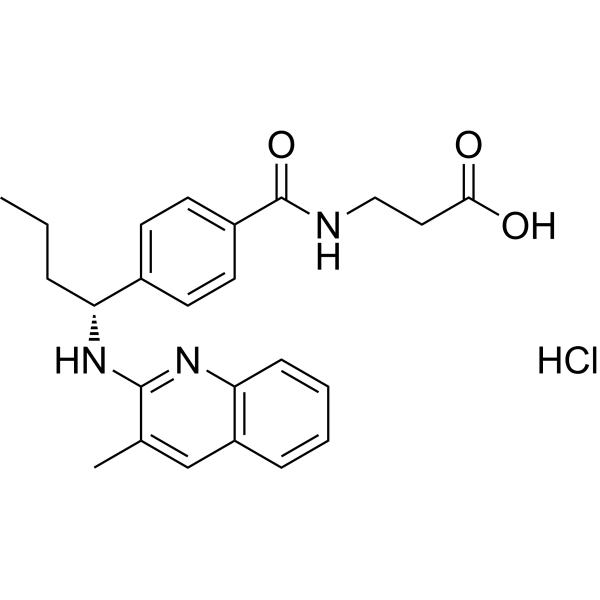 GCGR antagonist 3
GCGR antagonist 3
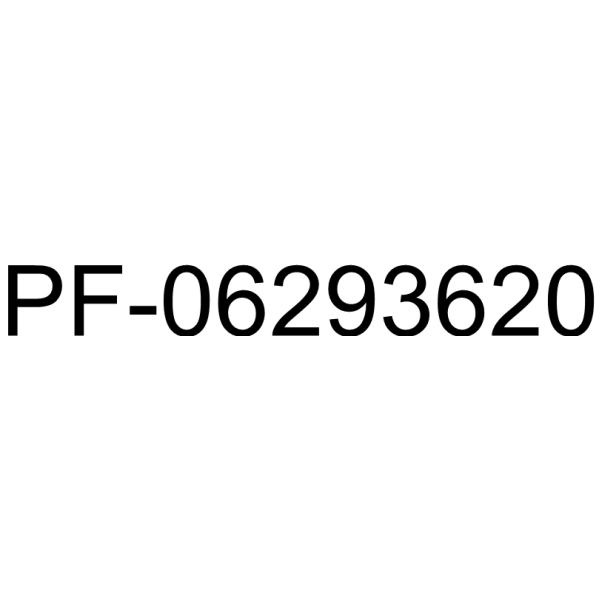 PF-06293620
PF-06293620
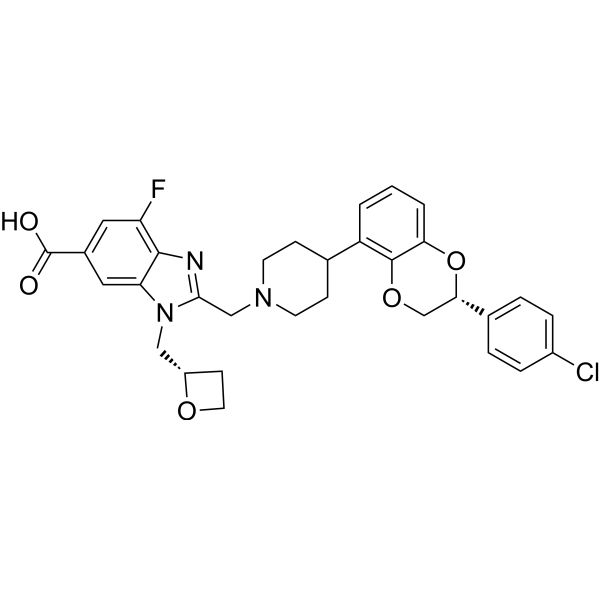 GLP-1R agonist 32
GLP-1R agonist 32
 Glucagon-like peptide 1 (1-37), human TFA
Glucagon-like peptide 1 (1-37), human TFA
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
