| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| Other Sizes |
|

| 靶点 |
YAP-TEAD (IC50 = 9 nM)[1]
IAG933 targets the protein-protein interaction between YAP1/WWTR1(TAZ) coactivators and all four TEAD transcription factor isoforms. By directly disrupting this interaction, IAG933 prevents TEAD from activating the transcription of genes involved in cell proliferation, survival, and oncogenesis. This mechanism of action is distinct from other TEAD inhibitors that target autopalmitoylation or the lipid pocket, offering a different approach to inhibiting Hippo pathway signaling. |
|---|---|
| 体外研究 (In Vitro) |
IAG933及其类似物是强效的首创选择性YAP-TEAD蛋白-蛋白相互作用阻断剂,具有进入临床试验的适宜特性。药理学阻断与所有四个TEAD旁系同源蛋白的相互作用,导致YAP从染色质中解离,并降低Hippo介导的转录和诱导细胞死亡。[2]
IAG933是一种直接的YAP-TEAD蛋白-蛋白相互作用阻断剂(PPIDs),靶向界面3,并于2021年进入临床试验[3]。 IAG933通过诱导细胞凋亡来增强对JDQ443的反应[2] 尽管选择性KRASG12C抑制剂对突变型癌症有影响,但其临床疗效通常不如RTK抑制剂显著。克服对KRASG12C抑制剂的耐药性仍然是一个挑战,促使人们开展正在进行的联合疗法临床试验[12]。与变构TEAD抑制剂的研究结果一致,IAG933与诺华KRASG12C抑制剂JDQ443在KRASG12C突变型非小细胞肺癌(NSCLC)和结直肠癌(CRC)细胞系中显示出显著的联合用药优势(图6a)。IAG933与其他JDQ443候选药物(如SHP2、MEK、ERK或PIKα抑制剂)相比,在不同细胞系中均表现出显著的生长抑制作用(扩展数据图9a)。在长期增殖实验中,我们观察到,当亚有效浓度的JDQ443与IAG933联合使用时,细胞生长受到持续且显著的抑制,而单独使用时,二者对细胞增殖的抑制作用较弱(扩展数据图9b)。在体内,持续地预先添加 IAG933 可加深 NCI-H2122 NSCLC 异种移植模型对 JDQ443 的反应(图 6b),并且这种组合优于 JDQ443 加 SHP2 抑制剂 TNO155(扩展数据图 9c)。在非小细胞肺癌(NSCLC)的PDX模型中也观察到了这种抗肿瘤联合效应,治疗结束后30天内未观察到肿瘤复发(图6c)。 IAG933特异性地阻断了YAP/TAZ共激活因子与所有四种TEAD亚型之间的相互作用,选择性地抑制了TEAD驱动的转录活性。这导致其产生抗癌作用。该化合物对YAP/TAZ-TEAD相互作用的选择性使其成为研究这种蛋白质-蛋白质相互作用在癌症和其他疾病中作用的宝贵工具。体外研究已证实其对TEAD依赖性转录和细胞增殖的影响。 |
| 体内研究 (In Vivo) |
IAG933 和 YTP-75 均表现出剂量依赖性的抗肿瘤疗效[2]
在小鼠 MSTO-211H 细胞来源的异种移植 (CDX) 模型中,采用灌胃法以 30 至 240 mg/kg 体重 (mg kg−1) 的单次剂量评估了 IAG933 的疗效。观察到剂量相关的血液暴露量,达到最大浓度的时间 (Tmax) 约为 1-2 小时,这与给药后约 2 小时开始的剂量/暴露依赖性 TEAD 靶基因抑制相一致(图 3a,b)。体内血液中靶基因抑制的 IC50 为 64 nM,略高于 MSTO-211H 细胞的体外 IC50 11-26 nM(图 1c)。利用原位胸膜 MSTO-211H 肿瘤中 TEAD 反应元件下的荧光素酶表达进行体内报告基因检测,结果显示单次注射 IAG933 后生物发光迅速且显著降低(图 3c),随后由于 IAG933 在小鼠体内的半衰期相对较短,数小时内恢复至基线水平。IAG933 类似物 YTP-75 也观察到类似的药代动力学/药效学结果(扩展数据图 5a、b),表明两种化合物均能快速且深入地抑制体内 TEAD 转录。 IAG933 在耐受剂量下可根除大鼠模型中的肿瘤[2] 为了将体内研究扩展到小鼠以外的其他动物,我们还在皮下 MSTO-211H 大鼠异种移植模型中评估了 IAG933。单次给药 IAG933 后靶基因抑制动力学与小鼠模型中观察到的结果相似(扩展数据图 7a、b),而 CCN2/ANKRD1/CCN1 的平均 IC50 值为 20 nM,约为体外 IC50 值的三分之一。每日给药 2 周后,在 10 mg kg⁻¹ 剂量下观察到肿瘤停滞,在 30 mg kg⁻¹ 剂量下,5 只动物中有 4 只观察到肿瘤完全消退(图 3g)。药物暴露量与剂量成正比,在 12 天的每日治疗中未检测到化合物蓄积(扩展数据图 7c)。未观察到体重减轻,且治疗耐受性良好。比较大鼠和小鼠模型反应曲线,确定大鼠每日一次 30 mg kg−1 的剂量与小鼠每日一次 240 mg kg−1 的剂量等效(扩展数据图 7d)。 IAG933 在间皮瘤和 Hippo 通路改变的异种移植瘤中的活性[2] 间皮瘤的发病机制通常涉及 Hippo 信号通路中抑癌基因的遗传改变,包括 NF2 和 LATS1/LATS2,估计在 32-50% 的病例中存在这种改变9,37-39。我们使用 9 种人源异种移植 (PDX) 小鼠模型,每日用 YTP-75 治疗,探索了 YTP 在不同间皮瘤遗传背景下的抗肿瘤疗效。在九个模型中的七个模型中观察到显著的肿瘤反应,其中三个NF2基因改变的模型出现深度肿瘤消退,而另外四个未报道Hippo通路改变的模型则出现持久的肿瘤停滞(图4a和扩展数据图7e)。有趣的是,两个未产生反应的肿瘤模型显示出TEAD靶基因最低的基础表达水平(图4a)。NF2突变在其他实体瘤中也以低发生率(~1-2%)被检测到9,38,40。为了探索YTP在这些病例中的活性,我们评估了IAG933在NF2基因改变的三阴性乳腺癌PDX模型(5938-HX)中的活性,以及YTP-75在NF2基因改变的肺癌CDX模型(NCI-H292)中的活性。两种模型均显示出对治疗的抗肿瘤反应,但5938-HX模型出现肿瘤消退(图4b),而NCI-H292模型对肿瘤生长的抑制作用较弱(图4c)。 IAG933联合治疗可提高RTK抑制剂的疗效[2] 此前研究表明,奥希替尼和VT104分别对EGFR和TEAD的共抑制可增强奥希替尼在非小细胞肺癌(NSCLC)模型中的肿瘤反应43。HER2阳性癌症和复发性癌症中已发现YAP活性升高3,8,而YAP-TEAD激活与曲妥珠单抗耐药性相关8,44。此外,近期数据表明,TEAD激活可在RTK抑制剂治疗下维持微小残留病灶6,43。因此,TEAD的共抑制可能对根除肿瘤至关重要。 RTK介导的癌症及肿瘤消除。 与此概念一致,IAG933联合奥希替尼显示出增强的抗肿瘤疗效,导致EGFR突变的NCI-H1975 NSCLC CDX模型中肿瘤快速消退(图5a)。此外,IAG933联合MET抑制剂卡马替尼在MET扩增的EBC-1肺癌CDX模型中诱导了显著的肿瘤缩小,而IAG933单药治疗则未观察到活性(图5b)。尽管IAG933在来自不同癌症适应症的七种HER2扩增细胞系中单药疗效有限,但IAG933与HER2抑制剂拉帕替尼联合用药显示出剂量依赖性的活性(图5c),并且在更长时间的体外研究中观察到治疗结束后仍存在持续的联合活性(图5d)。 (图5d)。体内实验表明,HER2扩增的NCI-N87胃癌异种移植模型在YTP-75联合曲妥珠单抗治疗下实现了肿瘤完全消退(图5e)。因此,在由不同RTK驱动的癌症模型中观察到了YTP的联合治疗获益,这表明它们具有共同的潜在机制,并为联合使用这些治疗药物提供了机会。 IAG933联合治疗在BRAFV600E突变肿瘤中显示出疗效[2] 由于BRAF的激活突变也会驱动致癌基因对MAPK通路的依赖,我们通过将IAG933与BRAF抑制剂达拉非尼、MEK1/MEK2抑制剂曲美替尼和/或ERK1/ERK2抑制剂LTT462联合使用,探索了YTP在BRAFV600E突变疾病中的联合治疗潜力。达拉非尼+IAG933、达拉非尼+LTT462+IAG933以及达拉非尼+曲美替尼+IAG933的组合在短期细胞活力检测中显示出疗效(图7a)。与TEAD活性在MAPK通路抑制中的适应性作用一致,在未联合YTP的情况下,达拉非尼+曲美替尼治疗后观察到TEAD反应基因表达增加,而联合使用IAG933类似物YTP-10抑制TEAD则可阻止这种增加(图7b)。在BRAFV600E突变的CRC CDX模型HT-29中,达拉非尼+LTT462+IAG933三联疗法比单药治疗显示出更强的抗肿瘤反应(图7c)。同样,达拉非尼+LTT462+IAG933三联疗法也显示出更强的抗肿瘤反应。在BRAFV600E突变型CRC异种移植模型5238-HX中,达拉非尼+曲美替尼+YTP-75的抗肿瘤活性强于达拉非尼+曲美替尼或达拉非尼+曲美替尼+西妥昔单抗,并在21天的研究期间实现了持续的肿瘤消退(图7d)。TEAD和RAF/MAPK阻断在非KRASG12C PDAC中的获益:除了临床可靶向的G12C变体外,KRAS驱动的肿瘤发生的治疗性抑制仍然具有挑战性52。为了治疗非KRASG12C突变型肿瘤,有效抑制下游RAF、MEK和/或ERK效应因子可能提供潜在的治疗选择。在此背景下,考虑到与突变特异性抑制剂联合用药所取得的令人鼓舞的结果(图66和图77),IAG933可能代表一种有前景的联合用药机会。 (图77和扩展数据图10)。我们在携带不同KRAS等位基因的PDAC细胞中验证了这一假设。在23种PDAC细胞系中,YTP-75与曲美替尼联合RAF抑制剂萘普生非尼联用显著增强了生长抑制作用(图8a),这与一项小鼠临床试验53的结果一致,该试验纳入了12个具有不同KRAS突变(7个G12D、2个G12V、2个Q61H和1个G12R)的PDAC PDX模型,其中8个模型(66%)在三联疗法下显示出肿瘤消退或接近停滞(图8b、c)。在SUIT-2 PDAC细胞中,通过基于荧光素酶的报告系统观察到曲美替尼+萘普生非尼可强烈诱导TEAD转录活性,而YTP-13联合治疗可抑制这种诱导作用(图8d)。在三种胰腺导管腺癌(PDAC)细胞系中,该组合已被证实能够抑制DUSP6和TEAD反应性基因ANKRD1的表达(图8e)。 IAG933口服有效,并在临床前模型中展现出抗癌作用。它能够抑制NCI-H2052细胞,可用于实体瘤侵袭和转移扩散的研究。该化合物的口服活性和首创的作用机制使其成为治疗Hippo通路驱动型癌症的潜在候选药物。体内研究表明,它能够抑制肿瘤生长并抑制TEAD驱动的转录活性。 |
| 酶活实验 |
表面等离子共振分析[2]
如前所述,使用人源TEAD1209–426、TEAD2221–447、TEAD3218–435和TEAD4217–434进行表面等离子共振分析测量。将四种N-生物素化的TEAD蛋白用AviTag标记并固定在传感器芯片上,在298 K下测量不同浓度YTP-3和YTP-32的结合情况。使用Biacore T200评估软件,采用1:1相互作用模型对数据进行全局拟合,以确定平衡状态下的解离常数(Kd)。 TR-FRET分析[2] 如前所述,在TR-FRET分析中测试了不同的化合物24。靶向肉豆蔻酸/棕榈酰口袋的脂质结合化合物(K-975 和 VT104)在 TR-FRET 检测中无活性,因为该检测中使用的 TEAD4 蛋白已完全酰化。 IAG933 的体外酶/受体结合(无细胞)检测通常包括蛋白质-蛋白质相互作用检测,以测量其破坏 YAP/TAZ-TEAD 相互作用的能力。将重组 YAP 或 TAZ 蛋白和 TEAD 蛋白与不同浓度的 IAG933 孵育,并使用表面等离子共振 (SPR)、AlphaScreen 或基于 ELISA 的方法测量相互作用。该化合物抑制 TEAD 驱动的转录活性的能力可以使用无细胞转录检测进行评估。 |
| 细胞实验 |
利用源自 SF-268 细胞的克隆进行集落形成试验的选择性评估[2]
对 SF-268 细胞系进行基因工程改造,并按如下方式建立了一个携带 TEAD1 双突变 (V406A/E408A) 的克隆。将 TEAD1 的靶向序列 (gtgcattcgctgtttcaaat) 克隆到 pNGx_006 载体 (pUC/ori,用于 tracrRNA/嵌合体的 U6 启动子,用于 SPyCas9 的 CMV 启动子和嘌呤霉素筛选) 中。将 1.5 μg pNGx_006_sgTEAD1 和 0.5 μg TEAD1V406A 和 TEAD1E408 的单链寡核苷酸(ttaacaggtggtaacaaacagggatacacaagaaactctactctgcatggcctgtgcattcgctgtttcaaatagtgaacacggagcacaacatcatatttacaggcttgtaaaggactg)电穿孔导入 SF-268 细胞 (2 × 10⁵),使用 Neon 转染系统,参数如下:电压 1300 V,脉冲宽度 20 ms,脉冲次数 2。嘌呤霉素筛选后,接种单克隆细胞,并通过 Sanger 测序进行鉴定。在克隆形成实验中,将 SF-268 克隆 18 和 23 以低密度(六孔板每孔 1000 个细胞)接种于处理前 24 小时。将测试化合物(IAG933)以10 μM的起始浓度进行五点三倍系列稀释,并分装至检测板中。DMSO用作对照,所有化合物处理孔中DMSO的含量均标准化为最高浓度。含化合物的培养基每周更换两次。在常规细胞培养条件(37 °C,5% CO2)下培养11天后,用3.7%甲醛固定细胞10分钟,并用结晶紫染色。 IAG933的体外细胞检测通常使用具有活跃YAP/TAZ-TEAD信号通路的癌细胞系,例如NCI-H2052细胞。用不同浓度的IAG933处理细胞,并使用TEAD反应性荧光素酶报告基因检测TEAD依赖性转录活性。细胞增殖、活力和侵袭能力采用CellTiter-Glo、MTT和侵袭实验等标准方法进行评估。化合物对TEAD靶基因表达的影响可通过qPCR或Western blot进行评估。 |
| 动物实验 |
动物实验[2]
大多数化合物均以指定剂量通过灌胃法给药,具体配方如下。IAG933 配制于 0.5% 甲基纤维素和 0.1% Tween-80 的 100 mM 磷酸盐缓冲液中(pH 值调至 8)。VT104 和 K-975 配制于 100% 甲基纤维素钠溶液中。YTP-75 配制于 30% PEG300 和 50 mM 醋酸盐缓冲液中(pH 值调至 5.5)。YTP-13 配制于 5% PEG300 和 50 mM 醋酸盐缓冲液中(pH 值调至 4.8)。LTT462、达拉非尼和曲美替尼配制于 20% MEPC4 水溶液中。 JDQ443、TNO155、奥希替尼和卡马替尼均配制成含0.5%甲基纤维素和0.1%吐温-80的水溶液。其他化合物通过腹腔注射给药。曲妥珠单抗和西妥昔单抗抗体以及MRTX1133化合物均配制成Dexolve溶液。 体内药效学研究[2] 动物按时间点和治疗分组,每组n=3-5只。采集血液、血浆和肿瘤样本用于药代动力学和药效学分析。血液样本在冰上采集,并储存于-20℃直至进一步处理。血浆和肿瘤样本在干冰上速冻,并储存于-80℃直至进一步处理。采用MSTO-211H STB-Luc原位胸膜间皮瘤肿瘤模型进行体内TEAD报告基因检测。每次测量前,小鼠均腹腔注射荧光素(150 mg kg−1)。20 分钟后,在小鼠清醒且固定时间不超过 1 分钟的条件下,使用 IVIS Spectrum 成像系统进行成像。 体内疗效研究[2] 当移植到侧腹的肿瘤体积至少达到 100 mm3 时开始治疗,并采用随机分组。疗效研究、肿瘤反应和复发情况均以治疗开始时的肿瘤体积为指标进行报告。对于异位模型的疗效研究,根据肿瘤体积将动物随机分组。肿瘤大小使用游标卡尺测量,并使用公式长 × 宽² × π/6 计算。为了评估疗效,有时会在实验结束时或达到最佳疗效时计算T/C百分比值,计算公式为(治疗组肿瘤体积变化量/对照组肿瘤体积变化量)× 100。对于肿瘤消退,则使用公式(治疗组肿瘤体积变化量/初始治疗组肿瘤体积)× 100来量化肿瘤反应。统计分析使用GraphPad Prism软件进行。对于胸膜原位移植模型的疗效研究,通过测量收集于EDTA抗凝微量采血管中的20 μl血液样本中的葡萄糖含量来评估活肿瘤负荷,样本储存于-20℃。向96孔白色板的每个孔中加入腔肠素(Nanolight)底物溶液(100 μl,100 mM),并加入5 μl血液,每个处理重复三次。使用 CentroXS LB960 发光仪测量生物发光,测量时间为 2 秒。 血液、血浆和肿瘤中化合物的生物分析检测方法[2] 采用超高效液相色谱-串联质谱 (UPLC-MS/MS) 法测定全血、血浆和组织中 IAG933 和 YTP-75 的浓度。冷冻组织样品根据制造商说明使用 CryoPrep 研磨成粉末,或使用 Fast Prep-24 系统在等体积的 HPLC 水中匀浆。将血液、血浆或组织样品(粉末或匀浆形式,约 25 mg,精确称重)与 25 µl 内标(1 µg ml⁻¹)混合,并加入 200 µl 乙腈进行提取以沉淀蛋白质。超声处理 5 分钟后,样品离心,取上清液 70 µl 与 60 µl HPLC 级水混合,然后取 5 µl 样品进行 UPLC-MS/MS 分析。样品注入反相色谱柱,流动相为甲酸水溶液和甲酸乙腈溶液。柱洗脱液直接导入三重四极杆质谱仪的离子源。采用电喷雾正离子多反应监测 (ESI-MRM) 模式进行 MS/MS 分析。使用非房室模型计算血管外给药的药代动力学 (PK) 参数,并根据平均值采用线性梯形法则进行拟合。 矩阵形式的组合分析[2] 使用 CellTiter-Glo 试剂通过 ATP 定量评估化合物组合对细胞增殖的影响。将细胞以每孔 300–700 个细胞的密度接种于白色壁、透明底的 384 孔板中,并在 37 °C 下孵育过夜。之后,使用 HP300 数字分液器以矩阵形式加入系列稀释的化合物或溶剂对照,每个处理均设置三个复孔。在化合物存在下孵育 5–7 天后,使用 CellTiter-Glo 试剂盒,按照供应商的说明监测细胞活力。数据使用内部开发的 Combination Analysis Module 程序进行分析。为了区分化合物的细胞毒性和细胞抑制作用,在化合物加入当天(第 0 天),还在单独的细胞培养板中评估了活细胞的数量,并用于计算细胞活力抑制程度。根据浓度矩阵中给定点的 CellTiter-Glo 信号值高于还是低于第 0 天(后者提示化合物处理导致细胞死亡),计算“生长抑制”(GI)值,计算方法如下:T < D0:GI = 100 × {1 – [(S – D0)/D0]};T ≥ D0:GI = 100 × [1 – (S – D0)/(V – D0)],其中 D0 为第 0 天,V 为溶剂对照,S 为信号值。该公式得到的数值范围为:0 表示与溶剂相比无化合物效应,100 表示生长停滞(即终点信号值等于第 0 天信号值),200 表示细胞完全死亡。在图6a中,IAG933采用三倍稀释法,NSCLC细胞系起始浓度为5.595 µM,CRC细胞系起始浓度为3 µM;JDQ443采用四倍稀释法,最高化合物浓度为1.6 µM。 IAG933的体内动物研究通常采用小鼠异种移植瘤模型。将携带源自YAP/TAZ-TEAD依赖性癌细胞系的皮下肿瘤的小鼠,通过口服不同剂量的IAG933进行治疗。监测肿瘤生长抑制情况,并在研究终点收集肿瘤组织,用于分析TEAD靶基因表达、增殖标志物和化合物暴露水平。该化合物对肿瘤侵袭和转移的影响也可在合适的模型中进行评估。 |
| 药代性质 (ADME/PK) |
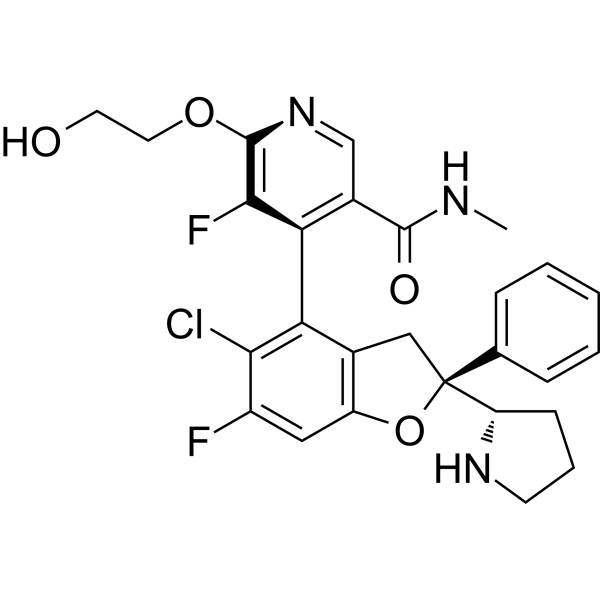
IAG933 是一种口服有效的化合物。其分子量为 529.96,分子式为 C27H26ClF2N3O4。现有文献中未详细描述其具体的药代动力学参数,例如半衰期、生物利用度、清除率和分布容积。该化合物的口服活性已通过临床前模型中的疗效得到证实。研究人员如需具体的药代动力学数据,应查阅相关文献。
|
| 毒性/毒理 (Toxicokinetics/TK) |
IAG933 的毒性数据尚未得到充分报道。作为一种研究用化合物,尚未进行全面的毒理学评估。该化合物仅供实验室研究使用,操作时应遵循标准安全预防措施。操作该化合物时应佩戴适当的个人防护装备。有关具体的安全和操作信息,请参阅材料安全数据表 (MSDS)。
|
| 参考文献 | |
| 其他信息 |
YAP-TEAD蛋白-蛋白相互作用介导YAP在Hippo信号通路下的致癌功能。目前,现有的YAP-TEAD药物通过结合TEAD的脂质口袋,间接靶向这一相互作用,导致全酶改变。然而,直接破坏YAP与TEAD之间界面的后果仍然不够清楚。本文介绍了IAG933及其类似物,作为首创的、高效的、选择性的YAP-TEAD蛋白-蛋白相互作用破坏剂,适合临床试验。药理学上阻断与所有四种TEAD同源物的相互作用导致YAP从染色质中解离,减少Hippo介导的转录并诱导细胞死亡。在动物模型中,在耐受剂量下,观察到Hippo驱动的间皮瘤异种移植和Hippo改变的非间皮瘤相关癌症模型中的肿瘤退缩。重要的是,IAG933的疗效还扩展到更广泛的肿瘤指示,如肺癌、胰腺癌和结直肠癌,并与RTK、KRAS突变选择性抑制剂和MAPK抑制剂联合使用,以实现更有效和持久的疗效。IAG933的临床评估正在进行中。[2] Hippo信号通路是一个高度保守的通路,在细胞增殖和凋亡的调节中发挥着重要作用。转录因子TEAD1-4和转录共调节因子YAP/TAZ是Hippo通路的下游效应因子,可以调节Hippo的生物功能。该通路的失调与肿瘤发生和获得性药物耐受性密切相关。YAP/TAZ-TEAD相互作用在癌症发展中的重要性日益突出,使其成为潜在的治疗靶点。在过去十年中,在阻断YAP/TAZ-TEAD相互作用作为有效癌症治疗方面取得了显著进展。该方法遵循以下轨迹:首先,设计了模仿YAP-TEAD蛋白-蛋白相互作用的肽类干扰剂(PPIDs),然后发现了别构小分子PPIDs,目前正在努力开发直接的小分子PPIDs。YAP和TEAD形成三个相互作用界面。界面2和3适合直接PPID设计。针对界面3的直接YAP-TEAD PPID(IAG933)于2021年进入临床试验。然而,与别构抑制剂的开发相比,战略性地设计针对TEAD界面2和3的有效小分子PPIDs通常更具挑战性。本综述重点关注直接表面干扰剂的开发,并探讨在开发高效YAP/TAZ-TEAD抑制剂用于癌症治疗中的挑战和机遇。[3]
IAG933是一种首创的、选择性的、口服有效的YAP1/WWTR1(TAZ)-panTEAD蛋白-蛋白相互作用抑制剂。它特异性地干扰YAP/TAZ共激活因子与所有四种TEAD亚型之间的相互作用,选择性抑制TEAD驱动的转录并诱导抗癌效果。该化合物在实体肿瘤的临床前模型中显示出疗效。IAG933尚未进入临床试验,也未获得治疗使用的批准。它仍然是研究Hippo信号通路和YAP/TAZ-TEAD生物学的研究工具。 |
| 分子式 |
C27H26CLF2N3O4
|
|---|---|
| 分子量 |
529.962852954865
|
| 精确质量 |
529.157
|
| CAS号 |
2714434-21-4
|
| PubChem CID |
156855755
|
| 外观&性状 |
White to off-white solid powder
|
| LogP |
3.7
|
| tPSA |
92.7Ų
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
8
|
| 可旋转键数目(RBC) |
7
|
| 重原子数目 |
37
|
| 分子复杂度/Complexity |
793
|
| 定义原子立体中心数目 |
2
|
| SMILES |
CNC(=O)C1=CN=C(C(=C1C2=C3C[C@@](OC3=CC(=C2Cl)F)([C@@H]4CCCN4)C5=CC=CC=C5)F)OCCO
|
| InChi Key |
HUVOYQMXUNTUAI-DCFHFQCYSA-N
|
| InChi Code |
InChI=1S/C27H26ClF2N3O4/c1-31-25(35)17-14-33-26(36-11-10-34)24(30)22(17)21-16-13-27(20-8-5-9-32-20,15-6-3-2-4-7-15)37-19(16)12-18(29)23(21)28/h2-4,6-7,12,14,20,32,34H,5,8-11,13H2,1H3,(H,31,35)/t20-,27-/m0/s1
|
| 化学名 |
4-[(2S)-5-chloro-6-fluoro-2-phenyl-2-[(2S)-pyrrolidin-2-yl]-3H-1-benzofuran-4-yl]-5-fluoro-6-(2-hydroxyethoxy)-N-methylpyridine-3-carboxamide
|
| 别名 |
YAP-TEAD-IN-3; IAG933; 2714434-21-4; IAG-933; NVP-IAG933; SCHEMBL23834952; GTPL13367; IAG933?;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 125 mg/mL (235.87 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8869 mL | 9.4347 mL | 18.8693 mL | |
| 5 mM | 0.3774 mL | 1.8869 mL | 3.7739 mL | |
| 10 mM | 0.1887 mL | 0.9435 mL | 1.8869 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT04857372
Conditions:Mesothelioma Verteporfin liposome
Verteporfin liposome
 HC278
HC278
 MY-1576
MY-1576
 YL-602
YL-602
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
