| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 1mg |
|
||
| 5mg |
|
||
| 10mg |
|
||
| Other Sizes |
|

| 靶点 |
BTK; Ikaros (IKZF1); Aiolos (IKZF3)
- Bruton's tyrosine kinase (BTK) (DC50 < 5 nM in multiple cancer cell lines and human PBMCs) [3] - Ikaros (IKZF1) and Aiolos (IKZF3) (DC50 of 54 nM and 25 nM, respectively, in primary human T cells) [3] |
|---|---|
| 体外研究 (In Vitro) |
- BTK降解:NX-2127在多种B细胞淋巴瘤细胞系(如TMD8、MEC1)和原代人B细胞中诱导剂量依赖性BTK降解,DC50值<5 nM。通过蛋白质印迹分析证实,24小时内BTK蛋白水平降低>80% [3]
- 类IMiD活性:该化合物通过降解原代人T细胞中的IKZF1和IKZF3表现出免疫调节作用,导致IL-2生成增加和T细胞活化,与来那度胺相当 [3] - 细胞增殖抑制:NX-2127有效抑制BTK依赖性ABC-DLBCL细胞(TMD8)的增殖,72小时后EC50 < 15 nM。值得注意的是,其对BTK C481S突变细胞的活性(EC50 < 30 nM)显著优于伊布替尼(>1 μM)[3] NX-2127 的 EC50 值 <30 nM,可抑制 BTK-C481S 突变 TMD8 细胞的增殖[1]。当暴露于 NX-2127 时,原代人类 T 细胞会产生更多的 IL-2 [1]。 |
| 体内研究 (In Vivo) |
- 肿瘤生长抑制:在小鼠异种移植模型(TMD8 WT和C481S突变)中,口服NX-2127导致剂量依赖性肿瘤生长抑制(TGI)。每日10 mg/kg剂量下,两种模型的TGI均超过80%,疗效优于伊布替尼 [3]
- 非人灵长类动物中的BTK降解:在食蟹猴中,每日一次口服给药(1–10 mg/kg)导致外周血单个核细胞(PBMCs)中BTK持续降解,水平至少24小时内抑制至<10%基线值 [3] 用 NX-2127(1 mg/kg;口服;每天一次,持续 14 天)治疗的食蟹猴显示出有效的 BTK 降解 [1]。口服 NX-2127 会导致血浆中的 BTK 在循环和脾 B 细胞中降解至基线水平的 10% 以下,且暴露量与剂量成比例[1]。在小鼠 WT TMD8 和 C481S 突变异种移植模型中,NX-2127 产生更强的肿瘤生长抑制 (TGI) [1]。 |
| 酶活实验 |
- BTK结合与降解实验:重组BTK蛋白与NX-2127和纯化的CRBN E3连接酶复合体孵育。通过SDS-PAGE和免疫印迹评估降解,显示剂量依赖性BTK泛素化和蛋白酶体降解。表面等离子体共振(SPR)证实BTK、NX-2127和CRBN的三元复合体形成,结合亲和力(KD)为0.8 nM [3]
- IKZF1/3降解实验:原代人T细胞用NX-2127处理,通过流式细胞术定量IKZF1/3水平。化合物在6小时内诱导两种蛋白快速降解,100 nM时效果最大 [3] 通过同源时间分辨荧光(HTRF)评估BTK降解细胞与化合物孵育4小时,并根据制造商的方案使用Cisbio Total BTK HTRF试剂盒(63ADK064PEG)测定BTK水平。HTRF比率使用以下方程式计算:[3] 除了有效的BTK降解外,NX-2127还通过设计CRBN结合线束具有IMiD样特性,该线束催化CRBN新底物Aiolos(IKZF3)和Ikaros(IKZF1)的降解。这种活性与IMiD药物来那度胺和泊马度胺的T细胞活化增加和抗肿瘤作用有关。在原代人类T细胞中,NX-2127分别以25 nM和54 nM的效力催化Aiolos和Ikaros的降解,其效力与来那度胺相似(分别为20 nM和343 nM)。与这种降解相对应,NX-2127以类似于来那度胺和泊马度胺的方式刺激T细胞活化,这是通过原代人T细胞中IL-2产量的增加来测量的。BTK降解的双重活性与NX-2127的免疫调节相结合,支持其治疗B细胞恶性肿瘤的发展。[1] |
| 细胞实验 |
人外周血单个核细胞中Aiolos和Ikaros降解的评估[3]
人外周血单核细胞(PBMCs)在37°C下用2000-0.00512 nM化合物处理24小时,然后使用Foxp3/转录因子固定/渗透试剂盒固定和渗透细胞。细胞用抗Aiolos、Ikaros、CD20和CD3的抗体染色。另外的样本用Aiolos和Ikaros同种型对照抗体染色作为染色对照。在Attune NxT声聚焦流式细胞仪上进行流式细胞术,并使用FlowJo(v10.5.3)和GraphPad Prism分析数据。对T细胞(CD3+CD20-)进行门控,计算Aiolos和Ikaros蛋白的几何平均荧光强度(MFI)。 活化T细胞分泌IL-2的定量[3] 对于T细胞活化,根据制造商的方案,通过免疫磁性阴性选择从白血病中分离出原代人类T细胞。T细胞用化合物预处理1小时,然后在化合物存在下用平板结合的抗CD3抗体(1µg OKT3/孔)和5µg/mL可溶性抗CD28(克隆28.2)刺激24小时。通过ELISA评估无细胞上清液中的总IL-2。 蛋白质组学检测[3] 用浓度为50nM的DMSO或降解剂处理人PBMC(每个样品200万个细胞)4小时。在-80°C下储存之前,测试细胞的存活率并用PBS洗涤。使用PreOmics iST 96x试剂盒进行蛋白质组学样品制备。随后,细胞被裂解,然后进行蛋白水解消化和清理。样品处理完成后,通过数据独立分析(DIA)通过液相色谱-串联质谱(LCMS/MS)进行数据采集。采集的DIA光谱使用Spectronaut(17.1.221229.55965)进行处理,使用无库S75专有directDIA算法和默认的BGS工厂设置。Q值截止值为1%的搜索结果(前体和蛋白质)被导出到Nurix开发的应用程序中,用于统计分析和可视化。 NX-2127是一种CTM,它包含一个连接到cereblon(CRBN)线束的BTK钩。NX-2127在多个癌症细胞系和人PBMC中以<5nM降解50%的细胞BTK(DC50)。BTK-CTMs损害BTK依赖性ABC-DLBCL细胞系TMD8的存活率(72小时后EC50:<15nM)。重要的是,NX-2127诱导细胞中突变的BTK-C481S降解,并比伊布替尼更有效地抑制BTK-C481 S突变TMD8细胞的增殖(NX-2127 EC50值<30 nM,而伊布替尼>1μM)[1]。 - B细胞中的BTK降解:从患者样本中分离的人CLL细胞用NX-2127(0.1–100 nM)处理。24小时后通过蛋白质印迹检测BTK蛋白水平,显示10 nM时降解>90%。降解与BTK突变状态无关(如C481S、L528W)[3] - T细胞活化实验:原代人T细胞与NX-2127和抗CD3/CD28磁珠共培养。ELISA定量IL-2分泌,显示剂量依赖性增加(EC50 10 nM),与来那度胺相当 [3] |
| 动物实验 |
体内药效学研究[3]
为进行药效学评估,将8-9周龄的雌性BALB/cJ小鼠或CD-1小鼠经口给予化合物或赋形剂。在指定时间点,通过尾静脉或心脏穿刺采集小鼠血液。裂解红细胞,并将剩余细胞用活/死细胞染料染色,然后固定,使用FoxP3/转录因子染色缓冲液套装进行透化处理,并用细胞表面标志物CD45、TCR β链、CD3和B220进行染色。为检测BTK胞内染色,使用抗BTK(克隆D3H5,Cell Signaling Technology,货号8547BC)一抗和AF647标记的抗兔二抗,并辅以小鼠FC阻断剂(BD553142)对细胞进行染色。使用 Attune NxT 声聚焦流式细胞仪进行流式细胞术分析,并使用 FlowJo 和 GraphPad Prism 分析数据。测量 B 细胞 (CD45+/B220+/TCR β-) 和 T 细胞 (CD45+/B220-/TCR β+) 的 BTK MFI,并使用以下公式计算降解率: 体内肿瘤移植 [3] 将 1 x 107 个 TMD8 细胞悬浮于 Hank's 平衡盐溶液 (HBSS) 和 Matrigel™ 中,比例为 1:1,皮下植入 7-8 周龄雌性 CB.17 SCID 小鼠(Fox Chase SCID®,CB17/Icr-Prkdcscid/IcrIcoCrl)的侧腹部。当肿瘤体积达到 60-175 mm³ 时,开始每日口服 NX-2127、伊布替尼或赋形剂。每周测量两次体重和肿瘤体积。肿瘤体积采用公式“长 x 宽² x 0.5”进行测量。肿瘤生长抑制率 (% TGI) 采用公式 [1 − (T − T0/C − T0)] × 100 计算,其中 T 和 C 分别代表治疗组 (T) 和对照组 (C) 的平均肿瘤大小,T0 指随机分组时的肿瘤大小。 犬样本的流式细胞术分析 [3] 比格犬 (Canis familiaris) 接受单次 NX-2127 给药,给药途径为静脉注射 1 mg/kg 或口服 10 mg/kg。动物在给药前和给药后间隔一段时间进行采血,并将血液样本收集到 Cyto-Chex BCT 管中。 非人灵长类动物样本的流式细胞术分析[3] 食蟹猴分别在 BASi 或 Charles River Laboratories(俄亥俄州阿什兰)每日口服一次赋形剂对照或 NRX-0492 或 NX-2127(10、30 或 100 mg/kg)。动物在第 1 天和第 14 天采血,并将血液样本收集到 Cyto-Chex BCT 管(Streck,213386)中。 小鼠口服 NX-2127 后,血浆中药物暴露量与剂量成正比,循环和脾脏 B 细胞中 BTK 降解至基线水平的 10% 以下。在野生型TMD8和C481S突变异种移植模型中,每日口服NX-2127均能显著抑制肿瘤生长(TGI),优于伊布替尼。NX-2127口服给药后,在食蟹猴体内也表现出对BTK的强效降解作用。在食蟹猴体内,每日一次口服给药14天后,即使剂量低至1 mg/kg,BTK水平也能被抑制至基线水平的10%以下。[1] NX-2127-001是一项首次人体试验,旨在评估NX-2127每日一次口服给药治疗复发/难治性B细胞恶性肿瘤成年患者的安全性、耐受性和初步疗效,该研究同时开展剂量递增(Ia期)和队列扩展(Ib期)研究。剂量递增将采用改良的斐波那契设计,每组纳入1例患者,之后根据方案规定的标准过渡到标准的3+3设计。1b期将设立至多5个扩展队列,招募慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(CLL/SLL)、弥漫性大B细胞淋巴瘤(DLBCL)、滤泡性淋巴瘤(FL)、套细胞淋巴瘤(MCL)、边缘区淋巴瘤(MZL)和华氏巨球蛋白血症(WM)患者。主要入组标准包括:既往接受过至少两线治疗(WM患者至少接受过一线治疗);可测量病灶;以及美国东部肿瘤协作组(ECOG)体能状态评分为0或1。预计将招募约130例患者(1a期30例,1b期100例),并接受治疗直至疾病进展或出现不可耐受的毒性反应。主要目标是评估NX-2127的安全性和耐受性,并确定最大耐受剂量(1a期),以及评估NX-2127在扩展队列中的早期临床活性(1b期)。该研究的 1a 期部分目前正在美国招募受试者。临床试验信息:NCT04830137。[2] - 小鼠异种移植模型:NX-2127 配制成 0.5% 甲基纤维素混悬液,并以口服方式(每日 10–30 mg/kg)给予携带 TMD8 肿瘤的 NSG 小鼠。每周测量两次肿瘤体积,所有剂量组均观察到显著的抑制作用。药代动力学分析显示,给药后 1–2 小时达到血浆峰浓度 (Cmax),支持每日一次给药方案。[3] - 食蟹猴研究:动物连续 14 天口服 NX-2127(1–10 mg/kg)。在预定的时间点采集血样,以评估外周血单核细胞 (PBMC) 中 BTK 的降解情况和血浆药物浓度。该化合物表现出线性药代动力学特征,末端半衰期约为24小时[3] |
| 药代性质 (ADME/PK) |
NX-2127 在不同物种中均表现出强大的体内靶点降解活性。静脉注射后,NX-2127 的清除率较低(表 4 和表 5)。小鼠口服 NX-2127 后,血浆暴露量呈剂量依赖性(图 3A),口服生物利用度中等,且小鼠血液中 BTK 的降解也呈剂量依赖性(图 3B)。小鼠单次口服 0.3、3、10 和 30 mg/kg 的 NX-2127 后,24 小时后循环 B 细胞中 BTK 水平分别降至基线的 81%、36%、21% 和 12%。除小鼠外,我们还在大鼠、犬和食蟹猴中进行了体内药代动力学-药效学 (PK-PD) 实验。NX-2127 在大鼠体内的清除率中等,口服生物利用度较低。 NX-2127 突出了靶向蛋白降解 (TPD) 催化机制的强大作用,尽管其生物利用度相对较低且清除率较高(表 5),但仍能有效促进犬和食蟹猴体内 BTK 的降解。犬和食蟹猴口服 10 mg/kg 剂量后,BTK 水平分别降至基线水平的 17% 和 9% [3]。吸收:小鼠口服生物利用度约为 60%,血药浓度峰值 (Cmax) 在 1-2 小时内达到。食蟹猴的绝对生物利用度约为 50%,支持每日一次给药 [3]。代谢:NX-2127 主要通过细胞色素 P450 酶 (CYP3A4) 在肝脏代谢,主要代谢产物为葡萄糖醛酸苷结合物。不到 5% 的剂量以原形经尿液排出 [3]
- 半衰期:小鼠和食蟹猴的终末半衰期分别约为 12 小时和 24 小时,这使得 BTK 能够持续降解 [3] |
| 毒性/毒理 (Toxicokinetics/TK) |
急性毒性:小鼠的半数致死剂量 (LD50) > 2000 mg/kg(口服),表明急性毒性较低。在食蟹猴中,剂量高达 10 mg/kg 时未观察到剂量限制性毒性 [3]
- 安全性:在 I 期临床试验中,NX-2127 的耐受性总体良好。常见不良事件包括中性粒细胞减少症、疲乏和腹泻,均为 1-2 级且可逆。未报告明显的 QT 间期延长或心血管毒性 [2] |
| 参考文献 |
|
| 其他信息 |
泽布鲁度胺是一种口服生物利用度高的嵌合靶向分子(CTM),可靶向降解布鲁顿酪氨酸激酶(BTK),具有潜在的免疫调节药物(IMiD)和抗肿瘤活性。泽布鲁度胺由一个通过连接子与 BTK 结合基团相连的 cereblon(CRBN)结合基团组成。给药后,泽布鲁度胺的 BTK 靶向基团可靶向并结合 BTK。结合后,CRBN 结合基团募集 CRBN(CRL4-CRBN E3 泛素连接酶复合物的组成部分)。这催化 BTK 的泛素化和蛋白酶体介导的降解,并阻止 B 细胞抗原受体(BCR)信号通路的激活。这既抑制了 B 细胞活化,也抑制了 BTK 介导的下游生存通路的激活。这导致过表达BTK的恶性B细胞生长受到抑制。此外,泽来布鲁胺可催化CRBN新底物Aiolos(IKZF3)和Ikaros(IKZF1)的降解,这两种转录因子均调控T细胞功能。这调节免疫系统的活性,增强T淋巴细胞的活化,从而增强T细胞介导的抗肿瘤作用。BTK是src相关BTK/Tec家族胞质酪氨酸激酶的成员,在B细胞恶性肿瘤中过表达;它在B淋巴细胞的发育、活化、信号传导、增殖和存活中发挥重要作用。CRBN是CRL4-CRBN E3泛素连接酶复合物的底物识别组分,在某些蛋白质的泛素化过程中起关键作用。与 BTK 抑制剂相比,泽来布胺可能克服与 BTK 抑制剂诱导的耐药突变相关的肿瘤耐药性。
- 作用机制:NX-2127 是一种双功能分子,可募集 CRBN E3 连接酶降解 BTK 和 IMiD 新底物(IKZF1/3)。这种双重活性可抑制 BCR 信号传导并增强 T 细胞介导的抗肿瘤免疫 [3] - 临床开发:在 I 期临床试验 (NCT04830137) 中,NX-2127 在复发/难治性 B 细胞恶性肿瘤(包括慢性淋巴细胞白血病 (CLL) 和套细胞淋巴瘤 (MCL))中显示出良好的疗效。在 27 例可评估的 CLL 患者中,总缓解率 (ORR) 为 40.7%(11 例部分缓解),缓解持续时间超过 12 个月 [2] - 克服耐药性:NX-2127 可有效降解赋予共价 BTK 抑制剂耐药性的 BTK 突变体(C481S、L528W),从而提供了一种与突变类型无关的治疗方法 [3] |
| 分子式 |
C39H45N9O5
|
|---|---|
| 分子量 |
719.831907987595
|
| 精确质量 |
719.354
|
| 元素分析 |
C, 65.07; H, 6.30; N, 17.51; O, 11.11
|
| CAS号 |
2416131-46-7
|
| PubChem CID |
146559796
|
| 外观&性状 |
Light yellow to yellow solid powder
|
| LogP |
3.7
|
| tPSA |
174
|
| 氢键供体(HBD)数目 |
3
|
| 氢键受体(HBA)数目 |
11
|
| 可旋转键数目(RBC) |
9
|
| 重原子数目 |
53
|
| 分子复杂度/Complexity |
1380
|
| 定义原子立体中心数目 |
1
|
| SMILES |
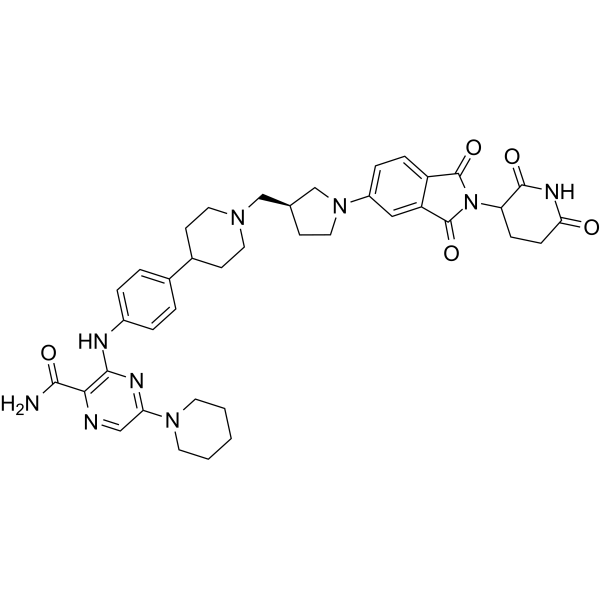
C1(C(N)=O)=NC=C(N2CCCCC2)N=C1NC1=CC=C(C2CCN(C[C@@H]3CCN(C4C=CC5=C(C=4)C(=O)N(C4CCC(=O)NC4=O)C5=O)C3)CC2)C=C1
|
| InChi Key |
XLWJWCMQMBVNSG-ACXKHFGCSA-N
|
| InChi Code |
InChI=1S/C39H45N9O5/c40-35(50)34-36(43-32(21-41-34)46-15-2-1-3-16-46)42-27-6-4-25(5-7-27)26-13-17-45(18-14-26)22-24-12-19-47(23-24)28-8-9-29-30(20-28)39(53)48(38(29)52)31-10-11-33(49)44-37(31)51/h4-9,20-21,24,26,31H,1-3,10-19,22-23H2,(H2,40,50)(H,42,43)(H,44,49,51)/t24-,31?/m0/s1
|
| 化学名 |
3-[4-[1-[[(3S)-1-[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-5-yl]pyrrolidin-3-yl]methyl]piperidin-4-yl]anilino]-5-piperidin-1-ylpyrazine-2-carboxamide
|
| 别名 |
NX-2127; 2416131-46-7; Zelebrudomide; 2-Pyrazinecarboxamide, 3-[[4-[1-[[(3S)-1-[2-(2,6-dioxo-3-piperidinyl)-2,3-dihydro-1,3-dioxo-1H-isoindol-5-yl]-3-pyrrolidinyl]methyl]-4-piperidinyl]phenyl]amino]-5-(1-piperidinyl)-; 3-[4-[1-[[(3S)-1-[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-5-yl]pyrrolidin-3-yl]methyl]piperidin-4-yl]anilino]-5-piperidin-1-ylpyrazine-2-carboxamide; NX2127; LSC67HA8DE; SCHEMBL21947733;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month 注意: 本产品在运输和储存过程中需避光。 |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
DMSO: 100 mg/mL (138.92 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.3892 mL | 6.9461 mL | 13.8922 mL | |
| 5 mM | 0.2778 mL | 1.3892 mL | 2.7784 mL | |
| 10 mM | 0.1389 mL | 0.6946 mL | 1.3892 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
Link: https://clinicaltrials.gov/ct2/show/NCT04830137
Conditions:Chronic Lymphocytic Leukemia (CLL)|Small Lymphocytic Lymphoma (SLL)|Waldenstrom Macroglobulinemia (WM)|Mantle Cell Lymphoma (MCL)|Marginal Zone Lymphoma (MZL)|Follicular Lymphoma (FL)|Diffuse Large B-cell Lymphoma (DLBCL)|Primary Central Nervous System Lymphoma (PCNSL)InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
