| 规格 | 价格 | |
|---|---|---|
| 500mg | ||
| 1g | ||
| Other Sizes |
| 靶点 |
Ki: 4.9 pM (hCD73)[1]
|
|---|---|
| 体外研究 (In Vitro) |
AB-680 ammium 可降低人、小鼠 CD8+ T 细胞和 hPBMC 中的 CD73,IC50 分别为 0.070、0.008、0.66 和 0.011 nM。它还在 CHO 细胞中抑制可溶性 hCD73,IC50 为 0.043 nM[1]。
|
| 体内研究 (In Vivo) |
基于AB680在体外的非凡效力和在临床前物种中良好的药代动力学特征,AB680被推进到健康志愿者的I期临床研究。这项安慰剂对照研究评估了AB680的安全性、耐受性和PK/PD特征。在这里,我们给出了AB680在人体内的初始药代动力学数据。AB680单次静脉给药在0.1 - 25mg范围内耐受性良好(6个队列)。所有治疗中出现的不良事件的严重程度均为轻度或中度,在这些剂量水平下没有明确的毒性模式。AB680在静脉输注16或25毫克30-60分钟后显示出低清除率和较长的半衰期(67-74小时)。AB680的人体PK谱与预期的Q2W给药计划一致,并验证了用于发现静脉给药的长效药物的设计策略。[1]
|
| 酶活实验 |
CYP抑制过程:[1]
测试化合物在体外评估其抑制细胞色素P450家族主要人类药物代谢酶的潜力。在0.1 mg/mL人肝微粒体蛋白悬浮液中,以0.1 M磷酸钾缓冲液(pH 7.4, NADPH 1 mM)和探针底物(非那西丁(CYP1A2),双氯芬酸(CYP2C9), s -美苯妥英(CYP2C19),右美沙芬(CYP2D6)和咪达唑仑(CYP3A4),分别在0至40µM的浓度范围内孵育测试化合物。每个底物在37°C下孵育5至20分钟,按照之前的实验验证定义。收集每个底物的样品,并与其他底物孵育的样品混合,用LC-MS/MS测定产物形成。采用变斜率(4参数)模型计算IC50值。以呋喃茶碱(1A2)、磺胺苯唑(2C9)、(+)- n -3-苄基硝基醇(2C19)、奎尼丁(2D6)、酮康唑(3A4)作为对照。
|
| 细胞实验 |
细胞人CD73测定。[1]
人CD73稳定CHO细胞系的生成与扩增Lake Pharma使用标准方案将携带人NT5E (CD73)基因的pcDNA3.1(+)载体转化CHO细胞,生成稳定的细胞系。在含有5 μg / mL purromycin和200 μg / mL潮霉素b的CD OptiCHO细胞培养基中进行抗生素选择,收集存活的CHO-CD73细胞池,在CD OptiCHO细胞培养基中7.5% DMSO冷冻。冷冻保存的细胞在37°C的水浴中通过搅拌小瓶解冻,直到细胞完全解冻。然后将细胞转移到15ml Falcon管中,以225 × g离心5分钟使细胞成球。将细胞颗粒重悬于添加2 mM谷氨酰胺的新鲜温CD OptiCHO生长培养基中,并转移到T175瓶中。两天后,在达到~80%的合流度(~ 2000万个细胞/烧瓶)时,细胞以1:3的比例分成三个新鲜的T175烧瓶。再过三天,将细胞转移到15 mL的Falcon试管中,在250 × g离心5分钟至成球。将细胞以每mL 300万个细胞的密度重新悬浮在CellBanker2低温保存培养基中,并将其放入低温小瓶中。细胞等分液在-80°C保存直至需要。
|
| 动物实验 |
Clinical Study[1]
The phase I clinical study was a first-in-human, double-blind, randomized, placebo-controlled combined single-ascending-dose (SAD) and multiple-ascending-dose (MAD) study to evaluate the safety, tolerability, PK, and potential PD effects of AB680 in healthy volunteers. Participants were randomly selected to receive AB680 (n = 6) or matching placebo (n = 2) in each of seven dosing cohorts in the SAD part and a single dose cohort in the MAD part. In the SAD part of the study, participants received a single iv infusion of 0.1, 0.6, 2, 4, 8, 16, 25 mg of AB680 or placebo. In the MAD part of the study, participants received iv infusion of 8 mg of AB680 or placebo once daily on 3 days (days 1, 8, and 15).
|
| 参考文献 | |
| 其他信息 |
Quemliclustat is a small molecule, competitive inhibitor of the ectoenzyme CD73 (cluster of differentiation 73; 5'-ecto-nucleotidase; 5'-NT; ecto-5'-nucleotidase), with potential immunomodulating and antineoplastic activities. Upon administration, quemliclustat targets and binds to CD73, leading to clustering of and internalization of CD73. This prevents CD73-mediated conversion of adenosine monophosphate (AMP) to adenosine and decreases the amount of free adenosine in the tumor microenvironment (TME). This prevents adenosine-mediated lymphocyte suppression and increases the activity of CD8-positive effector cells and natural killer (NK) cells. This also activates macrophages and reduces the activity of myeloid-derived suppressor cells (MDSCs) and regulatory T-lymphocytes (Tregs). By abrogating the inhibitory effect on the immune system and enhancing the cytotoxic T-cell-mediated immune response against cancer cells, tumor cell growth decreases. In addition, clustering and internalization of CD73 decreases the migration of cancer cells and prevents metastasis. CD73, a plasma membrane protein belonging to the 5'-nucleotidase (NTase) family, upregulated on a number of cancer cell types, catalyzes the conversion of extracellular nucleotides, such as AMP, to membrane-permeable nucleosides, such as adenosine; it plays a key role in adenosine-mediated immunosuppression within the TME.
Extracellular adenosine (ADO), present in high concentrations in the tumor microenvironment (TME), suppresses immune function via inhibition of T cell and NK cell activation. Intratumoral generation of ADO depends on the sequential catabolism of ATP by two ecto-nucleotidases, CD39 (ATP → AMP) and CD73 (AMP → ADO). Inhibition of CD73 eliminates a major pathway of ADO production in the TME and can reverse ADO-mediated immune suppression. Extensive interrogation of structure-activity relationships (SARs), structure-based drug design, and optimization of pharmacokinetic properties culminated in the discovery of AB680, a highly potent (Ki = 5 pM), reversible, and selective inhibitor of CD73. AB680 is further characterized by very low clearance and long half-lives across preclinical species, resulting in a PK profile suitable for long-acting parenteral administration. AB680 is currently being evaluated in phase 1 clinical trials. Initial data show AB680 is well tolerated and exhibits a pharmacokinetic profile suitable for biweekly (Q2W) iv-administration in human.[1] Introduction. Extracellular adenosine (ADO) suppresses immune function via inhibition of T cell and NK cell activation and is present in high concentrations in the tumor micro-environment (TME). Intra-tumoral generation of ADO depends on the sequential catabolism of ATP by two ecto-nucleotidases, CD39 (ATP→AMP) and CD73 (AMP→ADO). Inhibition of CD73 eliminates a major, non-redundant, pathway of ADO production in the TME and can reverse ADO-mediated immune suppression. Here we present the characterization of AB680, a novel, highly potent, reversible and selective small molecule inhibitor of CD73, currently in preclinical development as a potential anti-tumor agent. Methods. The potency of CD73 inhibitors was evaluated by measuring AMP hydrolysis by CHO-CD73 cells using a malachite green assay. Potency was also measured using human T-cells and soluble recombinant CD73. Selectivity against related ecto-nucleotidases was also assessed. In the presence of human serum, CD73 inhibition was measured by quantitation of AMP hydrolysis via luminescence. The ability of AB680 to reverse AMP-mediated immune suppression of human CD4+/CD8+ T cells was determined by adding exogenous AMP during T cell activation by anti-CD3/CD28/CD2 beads. The pharmacokinetic properties of AB680 were evaluated in multiple preclinical species. A projected human dosing schedule for AB680 was determined via allometric scaling. Results. AB680 is a highly potent, reversible and selective inhibitor of CD73 (IC50 < 0.01 nM on human CD8+ T-cells), which retains high potency in the presence of human serum. AB680 is > 10,000-fold selective against related ecto-nucleotidases and a large panel of unrelated enzymes, receptors, and ion channels. AB680 does not show significant inhibition of the major CYP450 isoforms or the hERG potassium channel. AB680 potently reverses AMP and ADO-mediated suppression of immune function in vitro. In the presence of high concentrations of AMP, AB680 robustly restored CD25 expression, IFN-γ production and proliferation of human CD4+ and CD8+ T-cells. The pharmacokinetics (PK) of AB680 were assessed in rodent and non-rodent species. The PK properties of AB680 are characterized by very low clearance and long half-lives in preclinical species, resulting in projected human PK properties suitable for intravenous (i.v.) dosing on a schedule consistent with typical mAb dosing cycles. High-dose infusions of AB680 in preclinical species were well tolerated. Conclusions. AB680 is a highly potent and selective small-molecule inhibitor of CD73 which can mitigate AMP and ADO-mediated tumor immunity by potently blocking the production of adenosine in the TME. AB680 exhibits a favorable projected human PK profile suitable for parental administration and is expected to enter clinical development in 2018.[2] |
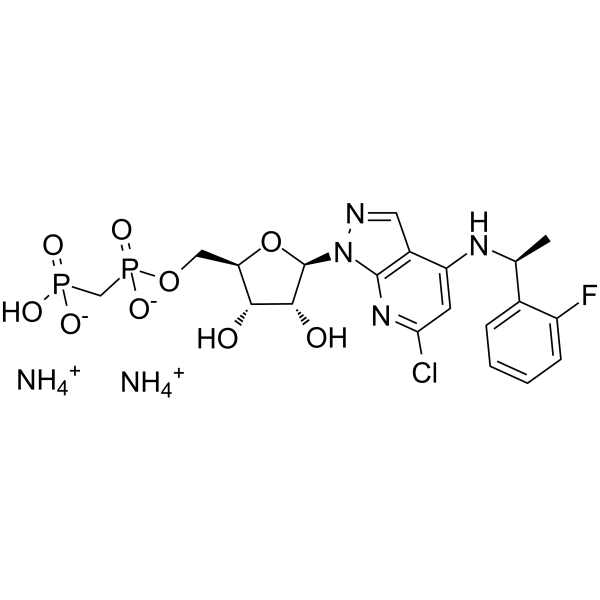
| 分子式 |
C20H30CLFN6O9P2
|
|---|---|
| 精确质量 |
614.12220
|
| 相关CAS号 |
AB-680;2105904-82-1
|
| PubChem CID |
162642790
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| tPSA |
204Ų
|
| InChi Key |
VVDIOJBTSCUIEW-AJLCLCFCSA-N
|
| InChi Code |
InChI=1S/C20H24ClFN4O9P2.2H3N/c1-10(11-4-2-3-5-13(11)22)24-14-6-16(21)25-19-12(14)7-23-26(19)20-18(28)17(27)15(35-20)8-34-37(32,33)9-36(29,30)31;;/h2-7,10,15,17-18,20,27-28H,8-9H2,1H3,(H,24,25)(H,32,33)(H2,29,30,31);2*1H3/t10-,15+,17+,18+,20+;;/m0../s1
|
| 化学名 |
diazanium;[(2R,3S,4R,5R)-5-[6-chloro-4-[[(1S)-1-(2-fluorophenyl)ethyl]amino]pyrazolo[3,4-b]pyridin-1-yl]-3,4-dihydroxyoxolan-2-yl]methoxy-[[hydroxy(oxido)phosphoryl]methyl]phosphinate
|
| 别名 |
AB-680 (ammonium); AB-680 ammonium
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 ZM522
ZM522
 CD73-IN-19
CD73-IN-19
 Mavrostobart
Mavrostobart
 Ansipastobart
Ansipastobart
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1
































 463611831
463611831
