| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| Other Sizes |
|

| 靶点 |
AAZ-A-154 targets 5-HT2 receptors (serotonin 2 receptors). It functions as a psychLight competitive antagonist and exhibits high selectivity for 5-HT2 receptors over dopamine, adrenergic, opioid, and other serotonin receptors. The compound shows negative ligand score (-31.7), indicating it is a non-hallucinogenic ligand of the 5-HT2AR. [1]
5-HT2A receptor (partial agonist, EC50 = 8.2 µM, efficacy = 17% span; Ki < 20 µM) [2][3] 5-HT2C receptor (activity observed; EC50 ~ 3.3 µM, Emax = 16%; Ki < 20 µM) [2][3] 5-HT2B receptor (full antagonist, no agonist activity; Ki < 20 µM) [2][3] 5-HT7 receptor (binding inhibition >50% at 10 µM; modest activity) [2][3] Serotonin transporter (SERT) (Ki < 20 µM; low potency inhibitor) [2][3] Sigma-1 receptor (Ki < 20 µM) [2][3] 5-HT1D receptor (modest activity) [3] 5-HT3 receptor (low potency inhibitor) [2][3] Monoamine oxidase A (MAO-A) (low potency inhibitor) [2][3] No detectable affinity for adrenergic or dopaminergic GPCRs, dopamine transporter (DAT), norepinephrine transporter (NET), or 5-HT1A receptors (EC50/IC50 > 30 µM). [2][3] |
|---|---|
| 体外研究 (In Vitro) |
AAZ-A-154(100 nM)促进培养的大鼠胚胎皮层神经元树突生长,增加树突分支复杂性,其程度与氯胺酮相当。这种促神经塑形作用可被5-HT2R拮抗剂酮色林(1 μM)阻断,表明AAZ-A-154通过激活5-HT2Rs触发树突生长。[1]
通过使用一组基于GPCR的传感器(多巴胺、肾上腺素能、阿片类和血清素受体)在激动剂和拮抗剂模式下测试,AAZ-A-154 对5-HT2受体表现出高选择性。[1] Schild回归分析显示,AAZ-A-154 是一种psychLight竞争性拮抗剂。[1] 在培养的胚胎大鼠皮层神经元中,Zalsupindole 以浓度依赖的方式促进神经发生。该效应已被三个独立实验室(Olson实验室、Neurofit、Cellectricon)重复验证。[2] Zalsupindole的促神经发生作用可被5-HT2拮抗剂酮色林和mTOR抑制剂雷帕霉素阻断,表明涉及5-HT2依赖的mTOR通路。[2][3] Zalsupindole不能激活基于5-HT2AR的生物传感器psychLight,无法诱导与致幻配体相关的构象变化。[2][3] 与致幻剂对照品不同,Zalsupindole在全身给药后不增加内侧前额叶或体感皮层中c-Fos(即早基因)的表达。[3] 在放射配体结合测定中(10 µM),Zalsupindole仅在5-HT7、5-HT2B和SERT处抑制>50%的放射配体结合。对肾上腺素能或多巴胺能GPCR、离子通道、DAT或NET无显著抑制。[2] 功能测定证实,Zalsupindole是5-HT2A的低效(EC50 = 8.2 µM)、低效能(span = 17%)部分激动剂,以及5-HT2B的完全拮抗剂。[2][3] |
| 体内研究 (In Vivo) |
AAZ-A-154(20 mg/kg,腹腔注射)在C57BL/6J小鼠的强迫游泳试验中产生快速(30分钟)和长效(1周)的抗抑郁样作用。该化合物减少了FST中的不动时间,这是一种已知的促神经塑形剂和抗抑郁药(如氯胺酮)通常产生的行为反应。[1]
在VMAT2杂合子小鼠(一种抑郁症模型)中,单次给予AAZ-A-154(15 mg/kg,腹腔注射)产生抗快感缺乏效应,治疗后的VMAT2-HET小鼠表现出与野生型对照无法区分的蔗糖偏好。该效应持续至少12天。蔗糖偏好的变化不能归因于液体摄入量的差异,因为两种基因型在整个实验期间饮用的液体体积相似,且AAZ-A-154不改变野生型动物的蔗糖偏好。[1] AAZ-A-154 即使在高达100 mg/kg(腹腔注射)的剂量下也未产生任何头部抽搐反应,证实了其非致幻性。然而,100 mg/kg的高剂量确实降低了小鼠的运动能力。[1] 在雄性大鼠中,单次腹腔注射Zalsupindole(3, 10, 30 mg/kg)后24小时,通过Golgi-Cox染色测得内侧前额叶皮层中树突棘密度呈剂量依赖性增加。效果与氯胺酮(10 mg/kg)和裸盖菇素(3 mg/kg)相当。对蘑菇状和丝状伪足状棘的作用最显著。[2] 在大鼠mPFC脑片中,腹腔注射后24小时,Zalsupindole(10 mg/kg)显著增加了自发兴奋性突触后电流的频率和幅度,表明功能可塑性增强。效应值大于氯胺酮或裸盖菇素,与DMT相当。[2] 在大鼠强迫游泳试验中,单次腹腔注射Zalsupindole(10 mg/kg)产生了快速(24小时内)且持续(至少7天)的抗抑郁样作用,减少不动时间,效果与氯胺酮(10 mg/kg)相似。[2][3] 在VMAT2杂合子小鼠(抑郁模型)中,单次Zalsupindole可缓解快感缺失(增加蔗糖偏好),效果持续>2周。[2] Zalsupindole在小鼠中(IP剂量高达30 mg/kg)不诱导头部抽搐反应,证实其无致幻潜力。[2][3] 在大鼠微透析实验中,10 mg/kg(IP)Zalsupindole短暂(2小时)增加血清素水平2倍,但不显著改变去甲肾上腺素、多巴胺、乙酰胆碱、GABA或谷氨酸。30 mg/kg时,增加血清素(4倍)和去甲肾上腺素(2倍)。未观察到谷氨酸爆发。[2][3] |
| 酶活实验 |
放射配体结合测定(CEREP):在10 µM浓度下测试Zalsupindole对55个CNS靶点的选择性。计算特异性放射配体结合的抑制百分比。抑制率>50%视为显著。[2]
5-HT2A和5-HT2C放射配体结合:竞争结合实验在96孔板中进行,包含结合缓冲液、膜提取物、[³H]-DOI示踪剂和测试化合物。非特异性结合由200倍过量血清素确定。样品室温孵育,过滤,洗涤,用MicroBeta计数器计数。使用Cheng-Prusoff方程计算Ki值。[2] IP-One HTRF测定(5-HT2A, 5-HT2C, 5-HT2B):表达人重组受体的CHO-K1细胞与测试化合物或参考激动剂(α-Me-5-HT)在37°C孵育60分钟。加入含IP1-d2和抗IP1-cryptate的裂解缓冲液后,测量荧光比率。5-HT2B使用类似方法。[2] MAO-A抑制测定:进行了人MAO-A酶学LeadHunter测定。[2] SERT结合测定:进行了人SERT拮抗剂放射配体LeadHunter测定。[2] Sigma-1受体结合测定:进行了人sigma-1激动剂放射配体LeadHunter测定。[2] GTPγS测定(5-HT1D):进行了基于HTRF的测定。[2] cAMP测定(5-HT7):进行了基于HTRF的测定。[2] 主要表征使用psychLight生物传感器和Schild回归分析。[1] |
| 细胞实验 |
培养皮层神经元的树突发生实验: 使用定时怀孕的Sprague Dawley大鼠(E18)获取皮层神经元。培养的神经元用AAZ-A-154(100 nM)或氯胺酮(1 μM,阳性对照)处理。为评估5-HT2Rs的作用,神经元与5-HT2R拮抗剂酮色林(1 μM)共处理。处理后,固定并染色神经元,使用Sholl分析分析树突分支复杂性以确定最大 crossings 数(Nmax)。AAZ-A-154增加Nmax,效果与氯胺酮相当,且该作用可被酮色林阻断。[1]
神经发生测定(Neurofit):从E17大鼠胚胎分离皮层神经元,接种于96孔板(10,000细胞/孔),用Zalsupindole处理72小时。细胞用多聚甲醛固定,透化,封闭,用抗β-III微管蛋白抗体和AF488二抗染色。细胞核用DAPI染色。使用高内涵图像分析仪成像和分析神经突网络。量化每个神经元的平均神经突数和总神经突长度。[2] 神经发生及阻断测定(Cellectricon):从E17.5大鼠胚胎分离皮层神经元,接种于384孔板(2500细胞/孔),培养72小时。使用HP D300数字分配器加入六个浓度的Zalsupindole(1:3稀释,起始10 µM)。阻断实验:在Zalsupindole前1小时加入酮色林(100 µM)或雷帕霉素(100 µM)。培养至第9天,细胞固定、封闭,用Hoechst(细胞核)和抗β-III微管蛋白(神经突)染色。使用高内涵成像仪和Harmony软件分析,量化总神经突长度。[2] |
| 动物实验 |
C57BL/6J小鼠强迫游泳试验: 使用雄性及雌性C57BL/6J小鼠(9-10周龄,每种性别每组6只)。在第一次FST前,连续3天每天对小鼠进行约1分钟的操作。未经药物处理的小鼠先进行一次预游泳以诱导抑郁样表型。次日,动物接受腹腔注射AAZ-A-154(20 mg/kg)、氯胺酮(3 mg/kg,阳性对照)或溶媒(生理盐水)。30分钟后,将小鼠放入装有30 cm 24±1°C水的透明有机玻璃圆筒中进行6分钟游泳测试。记录最后4分钟的不动时间。一周后重复FST以评估持续效应。所有FST均在上午8:00至下午1:00之间进行。[1]
头部抽搐反应和运动能力测定: 使用雄性和雌性C57BL/6J小鼠(约8周龄,每种处理2雄2雌,共4只)。使用0.9%生理盐水作为溶媒,腹腔注射给药(5 mL/kg)。注射后,将动物放入空笼中,录像记录HTR,随后由两名盲法观察者评分。使用自动化追踪软件评估运动能力。AAZ-A-154 在多个剂量(10、30、100 mg/kg)下进行测试,在任何剂量下均未产生HTR,但100 mg/kg剂量降低了运动能力。[1] VMAT2-HET小鼠蔗糖偏好试验: 实验前48小时将成年雄性和雌性野生型和VMAT2杂合子小鼠单独饲养,自由进食和饮水。暗周期开始前2小时,移除笼中水瓶。暗周期开始后1小时,将一对瓶子(水-水或水-蔗糖)放入笼中。让小鼠饮水2小时,然后移出瓶子并称重。每日重复此程序(水-水配对),直到连续3天小鼠饮水体积稳定。达到标准后,给予小鼠水-蔗糖配对。次日(第1天),小鼠接受急性注射AAZ-A-154(15 mg/kg,腹腔注射),5分钟后给予水-蔗糖配对。随后在第2天、第4天以及每4天间隔进行水-蔗糖配对。蔗糖偏好计算为(蔗糖消耗体积减去水消耗体积)除以总液体消耗体积。[1] 棘生成研究:雄性Sprague-Dawley大鼠(7周龄)单次腹腔注射Zalsupindole(3, 10, 30 mg/kg)、氯胺酮(10 mg/kg)、裸盖菇素(3 mg/kg)或溶媒(0.9%生理盐水),注射体积1 mL/kg。24小时后处死大鼠,取脑,进行Golgi-Cox染色(浸渍14天,切片100-120 µm)。使用Neurolucida软件量化mPFC中第5层锥体神经元的树突棘密度。[2] 电生理研究:雄性Sprague-Dawley大鼠(7-10周龄)腹腔注射Zalsupindole(10 mg/kg)、DMT(10 mg/kg)、氯胺酮(10 mg/kg)、裸盖菇素(1 mg/kg)或生理盐水。24小时后,麻醉大鼠并用冷ACSF灌注。制备含mPFC的脑切片(300 µm)。对第5层锥体神经元进行全细胞膜片钳记录,钳制电压-70 mV,记录sEPSC。使用MultiClamp 700B采集数据,pClamp分析。[2] 强迫游泳试验:雄性Sprague-Dawley大鼠(7-9周龄)在15分钟预游泳后,腹腔注射Zalsupindole(10 mg/kg)、氯胺酮(10 mg/kg)或生理盐水。24小时后以及第7天进行5分钟测试游泳。每5秒钟记录一次行为(不动、攀爬、游泳)。[2] 头部抽搐反应测定:雄性C57BL/6J小鼠(5-6周龄)腹腔注射裸盖菇素(1 mg/kg)、DOI(1 mg/kg)或Zalsupindole(10或30 mg/kg)。治疗后立即观察小鼠20分钟,由盲法测试者记录HTR事件(快速左右摇头)。[2] 微透析:雄性Sprague-Dawley大鼠(7-8周龄)在PFC植入I型微透析探针(AP +3.4 mm, L -0.8 mm, V -5.0 mm)。恢复后,用aCSF以1.5 µL/min灌注探针。收集基线样品后,大鼠腹腔注射Zalsupindole(3, 10, 30 mg/kg)。收集透析液4小时。通过HPLC-MS/MS分析神经递质水平(DA, NE, 5-HT, Glu, GABA, ACh)。[2] |
| 药代性质 (ADME/PK) |
在大鼠中,腹腔注射Zalsupindole(3, 10, 30 mg/kg)显示血浆和脑中快速且剂量成比例分布。半衰期约0.35小时,遵循一级动力学快速清除。脑浆比恒定在~10-25,表明高脑渗透性和低蓄积潜力。[2][3]
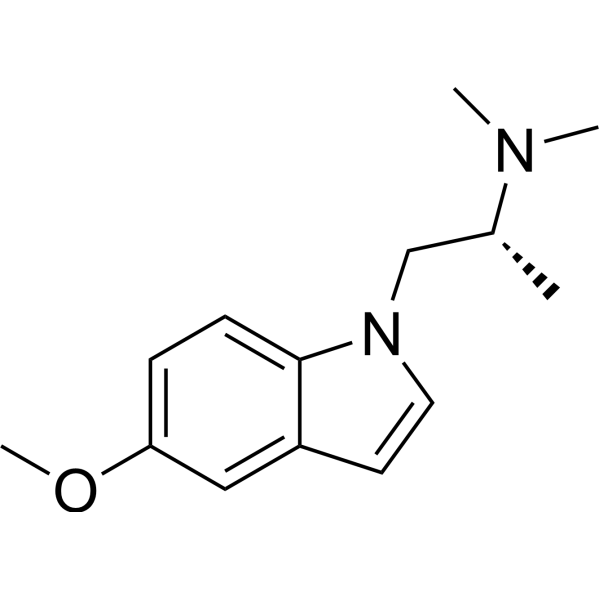
代谢:Zalsupindole经历去甲基化和氧化初级代谢,随后发生次级/三级葡萄糖醛酸化、羟基化和硫酸化。三种主要代谢物(M1, M2, M3)在神经突生长测定中无活性。[2][3] 在人体中(I期,口服给药,2-360 mg),Zalsupindole表现出线性和可预测的药代动力学、剂量比例吸收和良好的CNS渗透。食物摄入不显著改变吸收。[3] AAZ-A-154(化学名称:(R)-1-(5-甲氧基-1H-吲哚-1-基)-N,N-二甲基丙-2-胺富马酸盐)是一种迷幻剂的非致幻性类似物。其合成方法如下:将5-甲氧基吲哚溶于DMSO,加入(R)-1-氯-N,N-二甲基丙-2-胺盐酸盐、碘化钾和叔丁醇钾。反应混合物搅拌24小时,用NaOH(aq)稀释,DCM萃取,干燥,过滤,浓缩。产物通过闪式色谱纯化,溶于CHCl3,滴加到沸腾的富马酸THF溶液中,得到1:1富马酸盐(产率73%)。该化合物的配体评分为-31.7(负值,表明是非致幻性5-HT2AR配体)。AAZ-A-154 对5-HT2受体显示出高选择性。它在啮齿动物中单次给药后产生快速(30分钟)和长效(>2周)的有益行为效应。与另一种已知的非致幻性促神经塑形剂tabernanthalog相比,AAZ-A-154似乎更有效,同时产生更持久的抗抑郁效应。作者指出,未来应通过其他成熟 assays 验证其作用机制、脱靶效应、药代动力学、完整剂量反应研究、潜在毒性和疗效,以获得完整的药理学特征。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
在I期临床试验中(106名健康志愿者,口服剂量2-360 mg),Zalsupindole耐受性良好,无剂量限制性毒性、严重不良事件或治疗引发的幻觉。未报告精神模拟症状、解离或知觉紊乱。[3]
心血管安全性:Zalsupindole是5-HT2B受体的完全拮抗剂(无激动活性),表明心脏瓣膜病变风险低。I期试验中未观察到心率、血压或ECG参数变化。[2][3] 临床前或临床研究中未报告肝毒性或其他器官毒性。[2][3] 该化合物在高达100 mg/kg剂量下未产生头部抽搐反应,表明无致幻作用,但100 mg/kg的高剂量确实降低了运动能力。研究指出,未来应获得该化合物的完整药理学特征,包括潜在毒性。[1] |
| 参考文献 |
|
| 其他信息 |
Zalsupindole 也称为DLX-001和AAZ-A-154。由加州大学戴维斯分校Olson实验室使用psychLight生物传感器筛选非致幻5-HT2A激动剂时发现。[2][3]
作用机制:通过5-HT2A依赖的mTOR信号通路促进神经可塑性,不涉及Gq介导的致幻信号或谷氨酸爆发。低效能部分激动5-HT2A被认为是其产生治疗效果而无致幻作用的基础。[2][3] 适应症:正在开发用于重度抑郁症,并有可能用于PTSD、焦虑症、物质使用障碍和神经退行性疾病。[2][3] 临床状态:已完成I期(安全性、PK、EEG生物标志物)。在MDD中的Ib期研究显示,第8天MADRS评分降低约12分,持续至第36天。FDA已批准包含居家自我给药的II期试验设计。[3] 专利:物质组成专利US 11,254,640 B2涵盖Zalsupindole及其盐形式,保护期至少至2040年。WHO于2023年授予INN名称“Zalsupindole”。[3] AAZ-A-154(化学名称:(R)-1-(5-甲氧基-1H-吲哚-1-基)-N,N-二甲基丙-2-胺富马酸盐)是一种迷幻剂的非致幻性类似物。其合成方法如下:将5-甲氧基吲哚溶于DMSO,加入(R)-1-氯-N,N-二甲基丙-2-胺盐酸盐、碘化钾和叔丁醇钾。反应混合物搅拌24小时,用NaOH(aq)稀释,DCM萃取,干燥,过滤,浓缩。产物通过闪式色谱纯化,溶于CHCl3,滴加到沸腾的富马酸THF溶液中,得到1:1富马酸盐(产率73%)。该化合物的配体评分为-31.7(负值,表明是非致幻性5-HT2AR配体)。AAZ-A-154 对5-HT2受体显示出高选择性。它在啮齿动物中单次给药后产生快速(30分钟)和长效(>2周)的有益行为效应。与另一种已知的非致幻性促神经塑形剂tabernanthalog相比,AAZ-A-154似乎更有效,同时产生更持久的抗抑郁效应。作者指出,未来应通过其他成熟 assays 验证其作用机制、脱靶效应、药代动力学、完整剂量反应研究、潜在毒性和疗效,以获得完整的药理学特征。[1] |
| 分子式 |
C14H20N2O
|
|---|---|
| 分子量 |
232.32
|
| 精确质量 |
232.15756326
|
| 元素分析 |
C, 62.56; H, 7.88; Cl, 13.19; N, 10.42; O, 5.95
|
| CAS号 |
2481740-94-5
|
| 相关CAS号 |
2481740-94-5; 2930845-96-6 (HBr); 2930845-92-2 (benzoate); 2930845-89-7 (HCl); 2481740-95-6 (fumarate); 2930845-94-4; 2930846-03-8 (mesylate); 2481741-75-5 (S-isomer free base)
|
| PubChem CID |
154694212
|
| 外观&性状 |
Light yellow to yellow viscous liquid
|
| LogP |
2.4
|
| tPSA |
17.4 Ų
|
| 氢键供体(HBD)数目 |
0
|
| 氢键受体(HBA)数目 |
2
|
| 可旋转键数目(RBC) |
4
|
| 重原子数目 |
17
|
| 分子复杂度/Complexity |
244
|
| 定义原子立体中心数目 |
1
|
| SMILES |
C[C@H](CN1C=CC2=C1C=CC(=C2)OC)N(C)C
|
| InChi Key |
KHEUWLQKCXGVEL-LLVKDONJSA-N
|
| InChi Code |
InChI=1S/C14H20N2O/c1-11(15(2)3)10-16-8-7-12-9-13(17-4)5-6-14(12)16/h5-9,11H,10H2,1-4H3/t11-/m1/s1
|
| 化学名 |
(2R)-1-(5-methoxyindol-1-yl)-N,N-dimethylpropan-2-amine
|
| 别名 |
Zalsupindole; AAZ-A-154; DTXSID901336869; AAZA154; AAZ-A-154 HCl; AAZA-154 HCl; RefChem:1075133; DTXCID001767164; AAZ-A 154; AAZ-A154; AAZA 154; DLX-001 HCl; DLX 001 HCl; DLX001 HCl; 2481740-94-5;
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
Soluble in DMSO (>10 mM)
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.3044 mL | 21.5220 mL | 43.0441 mL | |
| 5 mM | 0.8609 mL | 4.3044 mL | 8.6088 mL | |
| 10 mM | 0.4304 mL | 2.1522 mL | 4.3044 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
 Antidepressant agent 10
Antidepressant agent 10
 Batoprazine
Batoprazine
 5-Hydroxy-6-methoxy (S)-duloxetine maleate
5-Hydroxy-6-methoxy (S)-duloxetine maleate
 Ifoxetine
Ifoxetine
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 COA
COA
 463611831
463611831
