| 规格 | 价格 | 库存 | 数量 |
|---|---|---|---|
| 5mg |
|
||
| 10mg |
|
||
| 25mg |
|
||
| 50mg |
|
||
| 100mg |
|
||
| 250mg |
|
||
| Other Sizes |
|

| 靶点 |
FTS also suppressed CER-induced ERK activation in the kidney. In vitro treatment of the established cell line, LLC-PK1 cells, with FTS significantly ameliorated CER-induced cell injury, as measured by lactate dehydrogenase (LDH) leakage. Our results, taken together with our previous report that MEK inhibitors ameliorated CER-induced renal cell injury and ERK activation induced by CER, suggest that FTS participates in protection from CER-induced nephrotoxicity by suppressing ERK activation induced by CER.[2]
|
|---|---|
| 体外研究 (In Vitro) |
FTS 还抑制了肾脏中 CER 诱导的 ERK 激活。用 FTS 体外治疗已建立的细胞系 LLC-PK1 细胞可显著改善 CER 诱导的细胞损伤,以乳酸脱氢酶 (LDH) 渗漏为衡量标准。我们的研究结果与我们之前的报告(MEK 抑制剂可改善 CER 诱导的肾细胞损伤和 CER 诱导的 ERK 激活)相结合,表明 FTS 通过抑制 CER 诱导的 ERK 激活参与了对 CER 诱导的肾毒性的保护。[2]
AZD9291 在携带EGFR突变的细胞系中表现出对EGFR磷酸化的强效抑制。在H1975细胞(DM: L858R/T790M)中,磷酸化抑制IC₅₀为15 nM。在PC9细胞(AM: 外显子19缺失)中,IC₅₀为17 nM。相比之下,在野生型EGFR细胞系如LoVo(IC₅₀ >480 nM)和Calu-3(抗增殖GI₅₀ = 264 nM)中活性弱得多,表明其对突变型EGFR的选择性显著高于野生型。[1] 在细胞抗增殖实验中,AZD9291 对突变EGFR驱动的细胞表现出强效的生长抑制。在H1975细胞(DM)中GI₅₀为24 nM,在PC9细胞(AM)中为23 nM,在Calu-3细胞(WT)中为264 nM。[1] 针对270种激酶组(Millipore panel)在1 µM浓度下的选择性分析表明,AZD9291 具有高度选择性,显著的抑制主要局限于少数酪氨酸激酶,包括EGFR突变体、IGF1R和INSR。[1] |
| 体内研究 (In Vivo) |
据报道,血清胸腺因子 (FTS) 是一种胸腺肽激素,可增加衰老加速小鼠的超氧化物歧化酶 (SOD) 水平。CER 可导致大鼠肾脏中的细胞外信号调节蛋白激酶 (ERK) 激活。因此,我们还研究了 FTS 是否对 CER 诱导的 ERK 激活有影响。电子显微镜照片显示,给雄性 Sprague-Dawley 大鼠静脉注射 CER (1.2 g/kg) 24 小时后,BUN 和血浆肌酐水平显著升高,尿葡萄糖和蛋白质排泄量显著增加,肌酐清除率降低,近端小管也出现显著病理变化。注射 CER 24 小时后,在从大鼠肾皮质制备的核级分中检测到磷酸化 ERK (pERK) 增加。用FTS(50 microg/kg,iv)预先处理大鼠可减轻CER引起的肾功能障碍和病理损伤。[2]
研究了血清胸腺因子(FTS)对BALB/cAJcl小鼠中脑心肌炎D型(EMC-D)病毒诱发的糖尿病和心肌炎的影响。用50或10微克FTS预先处理的小鼠感染10或10(3) PFU的EMC-D病毒。在接种10 PFU病毒的小鼠中,40%在感染后第14天(PID)患上糖尿病,而在感染前第2天和第1天用FTS(50微克/次给药)治疗的小鼠没有患上糖尿病。用FTS(10微克)预先处理的小鼠患上了糖尿病。在组织学观察中,未接受 FTS 治疗的患上糖尿病的小鼠在 19 PID 时,胰岛和心肌中的单核细胞出现严重坏死和炎症。然而,用 50 微克 FTS 进行预处理的小鼠表现出轻微的胰岛变性,而没有任何心肌炎症。此外,在未接受 FTS 治疗的小鼠中,免疫组织学染色显示胰岛素颗粒丢失。在接受 FTS 预处理的小鼠中,这种损失明显逆转,胰岛素颗粒基本保持完整。在伤后第 4、7 和 19 天,接受 FTS 预处理的小鼠胰腺中的病毒滴度与未接受处理的小鼠非常接近。然而,在接种了更高滴度 EMC-D 病毒(10(3) PFU)的小鼠中,50 微克 FTS 预处理并没有改变这些急性病理发展的进程(从伤后第 4 天开始观察到的糖尿病和心肌炎)。[3] 在小鼠异种移植模型中,口服给予AZD9291(5和10 mg/kg,每日一次,连续7天)在携带EGFR突变的模型中引起显著的肿瘤生长抑制和消退。在H1975(DM)模型中,10 mg/kg/天的剂量可诱导148%的肿瘤生长抑制(TGI)。在PC9(AM)模型中,10 mg/kg/天的剂量可诱导140%的TGI,即使在较低的5 mg/kg/天剂量下也能诱导122%的TGI。相比之下,它对表达野生型EGFR的A431异种移植模型影响极小。[1] 在携带H1975或PC9异种移植瘤的小鼠中,单次口服AZD9291(10 mg/kg)可导致作为药效学生物标志物的EGFR磷酸化(p-EGFR)被深刻且持续地抑制,观察到近完全抑制长达24小时。[1] |
| 酶活实验 |
AZD9291 对IGF1R和INSR的酶活性通过标准激酶实验测定。化合物的效力以IC₅₀值报告。[1]
使用Ionworks检测平台评估对hERG钾通道的抑制,测定抑制50%通道电流所需的浓度(IC₅₀)。[1] |
| 细胞实验 |
使用特定细胞系测量细胞EGFR磷酸化抑制活性(IC₅₀)。对于双突变EGFR,使用H1975人肺癌细胞系(携带L858R/T790M)。对于激活突变EGFR,使用PC9人肺癌细胞系(携带外显子19缺失)。对于野生型EGFR,使用LoVo人结肠癌细胞系或Calu-3人肺癌细胞系。细胞与化合物孵育2小时后,定量EGFR磷酸化水平,通常使用ELISA方法。IC₅₀值至少是两个独立测量的几何平均值。[1]
使用相同的细胞系组(H1975, PC9, Calu-3)评估细胞抗增殖活性(GI₅₀)。细胞暴露于化合物中,在一段时间后测量细胞活力/生长,以确定引起50%生长抑制的浓度。[1] 使用商业激酶组(Millipore 270激酶组)进行激酶选择性分析。化合物在1 µM单一浓度下测试,并测量每种激酶活性的抑制百分比。[1] |
| 动物实验 |
在小鼠异种移植模型中进行体内疗效研究时,将肿瘤细胞(H1975、PC9 或 A431)皮下植入雌性免疫缺陷(SCID 或裸鼠)小鼠体内。当肿瘤达到特定体积后,将小鼠随机分组。将 AZD9291(游离碱或甲磺酸盐)配制成 0.5% 羟丙基甲基纤维素 (HPMC) / 0.1% Tween 80 水溶液混悬液。每日一次 (qd) 口服给药,剂量为 5 或 10 mg/kg,持续 7 至 28 天。定期监测肿瘤体积和体重。[1] 在药效学 (PD) 研究中,携带 H1975 或 PC9 异种移植瘤的小鼠单次口服 AZD9291(10 mg/kg)。在不同时间点(例如,给药后 1、6、16、24 和 30 小时)采集肿瘤组织,并使用 ELISA 试剂盒检测肿瘤裂解液中磷酸化 EGFR (p-EGFR) 的水平。[1]
在大鼠毒理学和毒代动力学研究中,雄性 Han-Wistar 大鼠单次口服 AZD9291(50 或 200 mg/kg),以混悬液形式给药。在 24 小时内采集血样,以检测血浆药物浓度、葡萄糖水平和胰岛素水平。[1] |
| 药代性质 (ADME/PK) |
在临床前动物模型中,AZD9291 显示出中等至较高的体内清除率:静脉注射后,小鼠为 77 mL/min/kg,大鼠为 22 mL/min/kg,犬为 16 mL/min/kg。[1] 口服生物利用度存在个体差异,且与剂量相关。大鼠 5 mg/kg 剂量下的生物利用度为 13%;小鼠 10 mg/kg 剂量下的生物利用度为 31%;犬 5 mg/kg 剂量下的生物利用度为 67%。更高剂量导致药物暴露量增加,提示清除途径可能已饱和。[1] AZD9291 在体内会发生代谢。在啮齿动物和人体内,主要的循环代谢物是通过吲哚基团的N-去甲基化生成代谢物27 (M27),以及通过侧链的去烷基化生成代谢物28 (M28)。此外,还检测到一种少量的N-氧化物代谢物。[1]
在首次人体I期研究中,EGFR突变阳性非小细胞肺癌(NSCLC)患者每日口服一次20 mg甲磺酸盐。在血浆中检测到了AZD9291及其代谢物M27和M28,且患者间变异性较低。[1] AZD9291在pH 7.4时的logD值为3.4。其在pH 7.4时的水溶液溶解度为7 µM。血浆蛋白结合率高,在人血浆中的游离分数为2.9%。[1] |
| 毒性/毒理 (Toxicokinetics/TK) |
体外实验表明,AZD9291 对 hERG 钾通道的抑制 IC₅₀ 为 16.2 µM,提示其可能存在 QT 间期延长的风险,但风险相对较低。[1]
在一项大鼠毒理学研究中,早期先导化合物(20、21、22)单次口服高剂量(200 mg/kg)可强效抑制 IGF1R/INSR,导致显著的高血糖和高胰岛素血症。相比之下,相同剂量的 AZD9291 对血糖或胰岛素水平无显著影响,这与其较弱的 IGF1R/INSR 抑制作用相符。[1] 在豚鼠心血管安全性模型中,AZD9291 在游离血浆浓度显著高于预测的人体有效暴露量时,未引起显著的 QT 间期延长,这支持了其与其他先导化合物相比具有较高的安全性。 [1] AZD9291 的血浆蛋白结合率很高,通过平衡透析法测定,其在人血浆中的游离分数仅为 2.9%。[1] |
| 参考文献 |
|
| 其他信息 |
一种存在于正常血液中的胸腺依赖性九肽。它能刺激E玫瑰花结的形成,并被认为参与T细胞分化。
AZD9291(奥希替尼)是一种第三代不可逆EGFR酪氨酸激酶抑制剂(TKI),旨在克服非小细胞肺癌(NSCLC)患者中由T790M突变介导的耐药性,这些患者此前已接受过第一代EGFR TKI(例如吉非替尼、厄洛替尼)治疗。[1] 其作用机制涉及与突变EGFR的ATP结合位点中的半胱氨酸797残基共价结合,从而导致不可逆抑制。基于已发表的EGFR结构模型表明,其与铰链区(M793)的关键相互作用及其吲哚基团的取向至关重要。 [1] 一项针对T790M阳性、EGFR-TKI耐药的非小细胞肺癌(NSCLC)患者的I期临床试验(AURA)的初步临床数据显示,该药物疗效令人鼓舞。两名具有代表性的患者在接受每日一次20 mg的剂量治疗后,肿瘤显著缩小(根据RECIST 1.1标准缩小39-63%),临床症状也得到改善。治疗耐受性良好,早期患者中未出现严重皮疹,仅报告了轻微腹泻。[1] |
| 分子式 |
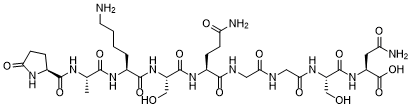
C33H54N12O15
|
|---|---|
| 分子量 |
858.86
|
| 精确质量 |
858.383
|
| CAS号 |
63958-90-7
|
| PubChem CID |
3085284
|
| 外观&性状 |
Typically exists as solid at room temperature
|
| 密度 |
1.417 g/cm33
|
| 沸点 |
1658.9ºC at 760 mmHg
|
| 闪点 |
957.1ºC
|
| 折射率 |
1.575
|
| LogP |
-10.9
|
| tPSA |
451.86
|
| 氢键供体(HBD)数目 |
15
|
| 氢键受体(HBA)数目 |
16
|
| 可旋转键数目(RBC) |
28
|
| 重原子数目 |
60
|
| 分子复杂度/Complexity |
1610
|
| 定义原子立体中心数目 |
7
|
| SMILES |
C[C@H](NC([C@@H]1CCC(N1)=O)=O)C(N[C@H](C(N[C@H](C(N[C@H](C(NCC(NCC(N[C@H](C(N[C@H](C(O)=O)CC(N)=O)=O)CO)=O)=O)=O)CCC(N)=O)=O)CO)=O)CCCCN)=O
|
| InChi Key |
LIFNDDBLJFPEAN-BPSSIEEOSA-N
|
| InChi Code |
InChI=1S/C33H54N12O15/c1-15(39-29(55)18-6-8-24(50)40-18)27(53)42-16(4-2-3-9-34)30(56)45-21(14-47)32(58)43-17(5-7-22(35)48)28(54)38-11-25(51)37-12-26(52)41-20(13-46)31(57)44-19(33(59)60)10-23(36)49/h15-21,46-47H,2-14,34H2,1H3,(H2,35,48)(H2,36,49)(H,37,51)(H,38,54)(H,39,55)(H,40,50)(H,41,52)(H,42,53)(H,43,58)(H,44,57)(H,45,56)(H,59,60)/t15-,16-,17-,18-,19-,20-,21-/m0/s1
|
| 化学名 |
(2S)-4-amino-2-[[(2S)-2-[[2-[[2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-5-oxopyrrolidine-2-carbonyl]amino]propanoyl]amino]hexanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]acetyl]amino]acetyl]amino]-3-hydroxypropanoyl]amino]-4-oxobutanoic acid
|
| 别名 |
thymulinSerum thymic factorNonathymulin
|
| HS Tariff Code |
2934.99.9001
|
| 存储方式 |
Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| 运输条件 |
Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs)
|
| 溶解度 (体外实验) |
May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples
|
|---|---|
| 溶解度 (体内实验) |
注意: 如下所列的是一些常用的体内动物实验溶解配方,主要用于溶解难溶或不溶于水的产品(水溶度<1 mg/mL)。 建议您先取少量样品进行尝试,如该配方可行,再根据实验需求增加样品量。
注射用配方
注射用配方1: DMSO : Tween 80: Saline = 10 : 5 : 85 (如: 100 μL DMSO → 50 μL Tween 80 → 850 μL Saline)(IP/IV/IM/SC等) *生理盐水/Saline的制备:将0.9g氯化钠/NaCl溶解在100 mL ddH ₂ O中,得到澄清溶液。 注射用配方 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (如: 100 μL DMSO → 400 μL PEG300 → 50 μL Tween 80 → 450 μL Saline) 注射用配方 3: DMSO : Corn oil = 10 : 90 (如: 100 μL DMSO → 900 μL Corn oil) 示例: 以注射用配方 3 (DMSO : Corn oil = 10 : 90) 为例说明, 如果要配制 1 mL 2.5 mg/mL的工作液, 您可以取 100 μL 25 mg/mL 澄清的 DMSO 储备液,加到 900 μL Corn oil/玉米油中, 混合均匀。 View More
注射用配方 4: DMSO : 20% SBE-β-CD in Saline = 10 : 90 [如:100 μL DMSO → 900 μL (20% SBE-β-CD in Saline)] 口服配方
口服配方 1: 悬浮于0.5% CMC Na (羧甲基纤维素钠) 口服配方 2: 悬浮于0.5% Carboxymethyl cellulose (羧甲基纤维素) 示例: 以口服配方 1 (悬浮于 0.5% CMC Na)为例说明, 如果要配制 100 mL 2.5 mg/mL 的工作液, 您可以先取0.5g CMC Na并将其溶解于100mL ddH2O中,得到0.5%CMC-Na澄清溶液;然后将250 mg待测化合物加到100 mL前述 0.5%CMC Na溶液中,得到悬浮液。 View More
口服配方 3: 溶解于 PEG400 (聚乙二醇400) 请根据您的实验动物和给药方式选择适当的溶解配方/方案: 1、请先配制澄清的储备液(如:用DMSO配置50 或 100 mg/mL母液(储备液)); 2、取适量母液,按从左到右的顺序依次添加助溶剂,澄清后再加入下一助溶剂。以 下列配方为例说明 (注意此配方只用于说明,并不一定代表此产品 的实际溶解配方): 10% DMSO → 40% PEG300 → 5% Tween-80 → 45% ddH2O (或 saline); 假设最终工作液的体积为 1 mL, 浓度为5 mg/mL: 取 100 μL 50 mg/mL 的澄清 DMSO 储备液加到 400 μL PEG300 中,混合均匀/澄清;向上述体系中加入50 μL Tween-80,混合均匀/澄清;然后继续加入450 μL ddH2O (或 saline)定容至 1 mL; 3、溶剂前显示的百分比是指该溶剂在最终溶液/工作液中的体积所占比例; 4、 如产品在配制过程中出现沉淀/析出,可通过加热(≤50℃)或超声的方式助溶; 5、为保证最佳实验结果,工作液请现配现用! 6、如不确定怎么将母液配置成体内动物实验的工作液,请查看说明书或联系我们; 7、 以上所有助溶剂都可在 Invivochem.cn网站购买。 |
| 制备储备液 | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.1643 mL | 5.8217 mL | 11.6433 mL | |
| 5 mM | 0.2329 mL | 1.1643 mL | 2.3287 mL | |
| 10 mM | 0.1164 mL | 0.5822 mL | 1.1643 mL |
1、根据实验需要选择合适的溶剂配制储备液 (母液):对于大多数产品,InvivoChem推荐用DMSO配置母液 (比如:5、10、20mM或者10、20、50 mg/mL浓度),个别水溶性高的产品可直接溶于水。产品在DMSO 、水或其他溶剂中的具体溶解度详见上”溶解度 (体外)”部分;
2、如果您找不到您想要的溶解度信息,或者很难将产品溶解在溶液中,请联系我们;
3、建议使用下列计算器进行相关计算(摩尔浓度计算器、稀释计算器、分子量计算器、重组计算器等);
4、母液配好之后,将其分装到常规用量,并储存在-20°C或-80°C,尽量减少反复冻融循环。
计算结果:
工作液浓度: mg/mL;
DMSO母液配制方法: mg 药物溶于 μL DMSO溶液(母液浓度 mg/mL)。如该浓度超过该批次药物DMSO溶解度,请首先与我们联系。
体内配方配制方法:取 μL DMSO母液,加入 μL PEG300,混匀澄清后加入μL Tween 80,混匀澄清后加入 μL ddH2O,混匀澄清。
(1) 请确保溶液澄清之后,再加入下一种溶剂 (助溶剂) 。可利用涡旋、超声或水浴加热等方法助溶;
(2) 一定要按顺序加入溶剂 (助溶剂) 。
InvivoChem的所有产品仅用于作科学研究,不面向患者销售
Copyright 2020 InvivoChem LLC | All Rights Reserved 粤ICP备20063088号-1































 463611831
463611831
